Journal of Biosciences and Medicines
Vol.05 No.01(2017), Article ID:73247,10 pages
10.4236/jbm.2017.51001
Forskolin Modulates the Inhibitory Effect of C-Type Natriuretic Peptide on Hypoxia-Induced Atrial Dynamics and Hypoxia Inducible Factor 1 Alpha Activity
Chengming Guan1, Yanan Jia1, Chaochao Bian2, Bo Zhang2, Dazhi Ding1*, Xun Cui2,3,4*
1Institue of Clinical Medicine, Yanbian University, Yanji, China
2Department of Physiology, School of Medicine, Yanbian University, Yanji, China
3Key Laboratory of Organism Functional Factors of the Changbai Mountain, Ministry of Education, Yanbian University, Yanji, China
4Cellular Function Research Center, Yanbian University, Yanji, China

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 1, 2016; Accepted: December 31, 2016; Published: January 3, 2017
ABSTRACT
Our study investigated effects of C-type natriuretic peptide (CNP) on atrial dynamics and hypoxia inducible factor 1 alpha (HIF-1α) activity in perfused beating rat atria, under hypoxic conditions. Hypoxia significantly increased the levels of HIF-1α, concomitant with decreased trial dynamics. CNP (0.1 µmol/L) further decreased atrial dynamics under hypoxia and suppressed hypoxia-induced stimulation of HIF-1α expression. An adenylylcyclase (AC) activator, forskolin (0.1 µmol/L), significantly up-regulated atrial phosphodiesterase subtype 3A (PDE 3A) protein without affecting hypoxia-induced dynamics. In the presence of forskolin, the inhibitory effects of CNP on hypoxia- induced atrial dynamics and HIF-1α levels were significantly attenuated. Fors- kolin also prevented hypoxia-induced downregulation of PDE3A protein. These findings suggested that CNP inhibited atrial dynamics and HIF-1α activity in the isolated perfused beating rat atria under hypoxic conditions. Furthermore, both effects were modulated by the AC activator forskolin, through activation of CNP-PDE 3A signaling.
Keywords:
C-Type Natriuretic Peptide, Hypoxia Inducible Factor-1α, Phosphodiesterase, Adenylyl Cyclase, Forskolin

1. Introduction
Hypoxia is a common phenomenon in most cardiovascular diseases, including coronary artery disease, heart failure, myocardial hypertrophy and pulmonary hypertension [1] [2] [3] . Hypoxia-inducible factor-1 (HIF-1) is a heterodimeric transcription factor that plays a major role in cellular adaptation to hypoxia [4] . It is composed of HIF-1α and HIF-1β subunits, and its activity is dependent on stability of the α-subunit [5] [6] . It was reported that cyclic adenosine monophosphate (cAMP)-dependent protein kinase (protein kinase A, PKA) phospho- rylated Thr63 and Ser692 on HIF-1α in vitro, enhancing its transcriptional activity and increasing target gene expression of rat cardiomyocytes. PKA also stimulated binding of the coactivator p300 to HIF-1α, enhancing its transcriptional activity while counteracting inhibition by asparaginyl hydroxylation of the association of p300 with HIF-1α [7] [8] . Thus, cAMP promotes HIF-1 transcriptional activity and increases HIF-1α protein levels through PKA activation, exerting physiological and pathophysiological effects on the myocardium.
As an endocrine gland, the heart produces and secretes natriuretic peptides (NPs), such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide (CNP). Hypoxia potently stimulated cardiac ANP and BNP secretion [9] [10] . ANP and BNP conferred resistance in the ischemic heart to hypoxia and myocardial cell damage, resulting in cellular adaptation to hypoxia and cardioprotection, through activation of cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) signaling [11] [12] . Several studies demonstrated that ANP and BNP markedly down-regulated HIF-1α during renal ischemia/reperfusion (I/R) injury in mice [13] [14] . However, effect of CNP on HIF-1α regulation in the atrium is unclear. Our study, therefore, investigated effects of CNP on hypoxia-induced HIF-1α levels in isolated beating rat atria. We also evaluated effects of the adenylyl cyclase (AC) activator, forskolin, on regulation of hypoxia-induced HIF-1α levels by CNP.
2. Materials and Methods
2.1. Preparation of Perfused Beating Rat Atria
Sprague-Dawley (SD) rats of both sexes were used, with mean weights of 250 - 300 g. Isolated perfused beating left atria were prepared as previously described [15] [16] . Soon after setting up each perfused atrium, transmural electrical field stimulation with a luminal electrode was started at 1.5 Hz (0.3 ms, 30 - 40 V), and the atrium was perfused with HEPES buffer solution using a peristaltic pump (1 mL/min), allowing atrial pacing for measurement of changes in atrial pulse pressure. The perfused atrium was supplied with sufficient oxygen during the entire process. The HEPES buffer contained (in mmol/L) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 10 glucose, and 10 HEPES (pH 7.4 with NaOH), as well as 0.1% bovine serum albumin.
2.2. Hypoxic Atrial Model Preparation
The hypoxic atrial model was prepared as previously described [9] . Briefly, the atrial O2 was replaced by N2 gas and the normal HEPES buffer was replaced with N2-saturated HEPES buffer.
Intra-atrial pressure was recorded using a Physiograph (Power Lab 2/20) via a pressure transducer (Statham P23Db, Oxnard, CA, USA) and pulse pressure was calculated by the difference between systolic and diastolic pressures. Pulse pressures were expressed as cm H2O.
Changes in atrial pulse pressure (fold) = (value of pulse pressure − mean basal value of pulse pressure)/mean basal value of pulse pressure.
2.3. Experimental Protocol
Each atrium was perfused for 60 min to stabilize atrial dynamics and then the control cycle (12 min as an experimental cycle) was followed by infusion of hypoxic buffer for four cycles, monitoring changes in atrial dynamics. For western blot analysis, immediately after perfusion, the atrial tissue was frozen and stored at −80˚C until analyzed.
To investigate effects of CNP and forskolin on hypoxia-induced atrial dynamics, one cycle of hypoxia after the control was followed by three cycles of infused treatment agent plus hypoxia. The treatment agents used were CNP (0.1 µmol/L) and forskolin (0.1 µmol/L). In the control group, vehicle was introduced instead of treatment agent. Values obtained during the periods corresponding to control and experimental observations were compared.
2.4. Western Blot Analysis
Proteins derived from left atrial tissue were analyzed by western blotting. Atrial tissues were homogenized in radio-immunoprecipitation assay lysis buffer (Solarbio institute of Biotechnology, Shanghai, China), and protein concentrations were determined with a Bradford protein assay kit. Solubilized proteins were denatured in Lane Maker Loading buffer and proteins separated by 10% or 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Protein bands were then transferred to polyvinylidene difluoride filter membranes (Beyotime Institute of Biotechnology, China). Each membrane was blocked with a 5% skim milk in phosphate buffer (PBST) solution at room temperature. After 2 h the membranes were incubated with the appropriate primary antibodies, overnight at 4˚C. The primary antibodies used were anti-phosphodiesterase subtype 3A (PDE3A, 1:1000, Abcam Shanghai, Shanghai, China) or anti-HIF-1α (1:1000, Abcam Shanghai), using, rabbit polyclonal β-actin (1:1000; Com Win Biotech, Beijing, China) as a loading control for all lanes. The membranes were then washed and incubated with secondary antibodies (1:2000) at room temperature for 2 h. After washing membranes thoroughly with PBST, stained bands were visualized by the ECL method (ECL Western Blot Kit, Com Win Biotech) and band densities quantified using Image J software (National Institutes of Health, Bethesda, MD, USA).
2.5. Statistical Analysis
The significance of differences among values was determined by one-way ANO- VA followed by Dunnett’s multiple comparison test. An unpaired t-test was also applied. Statistical significance was defined as P < 0.05. All data were presented as means ± SEM.
3. Results
3.1. Effect of CNP on Hypoxia-Induced Atrial Dynamics
As shown in Figure 1, hypoxia significantly decreased pulse pressure in isolated perfused beating rat atria (P < 0.05 vs. control, (a)). CNP also substantially
 (a)
(a)
 (b)
(b)
 (c)
(c)
Figure 1. Effect of CNP (0.1 µmol/L) on hypoxia-induced pulse pre- ssure (PP) in isolated perfused rat atria. (a) hypoxia-induced PP; (b) and (c) CNP modulated PP (data in (c) were derived from (a) and (b) control as well as the last cycle of the experimental period). Data were expressed as mean ± SEM, n = 6. *P < 0.05 vs. control; #P < 0.05 vs. hypoxia.
decreased pulse pressure in the hypoxic atria (P < 0.05 vs. control, (b)), a net effect that was greater than that of hypoxia alone (P < 0.05 vs. hypoxia alone, (c)). These data indicated that CNP had a negative inotropic effect in the hypoxic atrium.
3.2. Effects of Forskolin on CNP-Induced Suppression of Hypoxic Atrial Pulse Pressure
To determined effects of an AC activator, forskolin, on regulation of hypoxia- induced atrial dynamics by CNP, a series of experiments were performed with this agent in the perfused beating rat atria. As shown in Figure 2, forskolin did not affect hypoxia-induced atrial pulse pressure (P < 0.05 vs. control; P > 0.05 vs. hypoxia). In contrast, the AC activator dramatically attenuated the inhibitory effects of CNP on hypoxia-induced pulse pressure (P < 0.05 vs. hypoxia; P < 0.05 vs. CNP; P < 0.05 vs. forskolin). These results suggested that forskolin reversed the inhibitory effects of CNP on atrial dynamics in the beating hypoxic atrium.
3.3. Effects of Forskolin and CNP on Atrial PDE3A Levels under Hypoxia
PDE 3A regulates intracellular cAMP levels, so we examined levels of this protein to investigate the mechanism of forskolin-mediated reversal of inhibition by CNP of hypoxia-induced atrial pulse pressure. Atrial PDE3A levels were determined by western blotting in hypoxic beating atria that had been treated with or without forskolin and/or CNP. Forskolin significantly up-regulated atrial PDE 3A protein levels under hypoxic conditions (P < 0.05 vs. control group; P < 0.05 vs. hypoxia group, Figure 3). There were no significant changes in PDE 3A levels with hypoxia alone or with hypoxia plus CNP. However, in the presence of forskolin, CNP dramatically suppressed levels of PDE 3A in hypoxic atria (P < 0.05 compared with all other groups, Figure 3). This indicated that CNP inhibited forskolin-induced PDE3A activity activation in hypoxic atria.
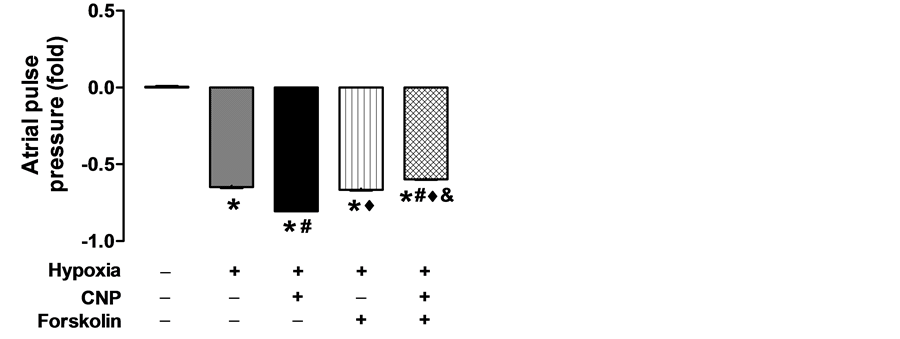
Figure 2. Effect of forskolin (0.1 µmol/L), an activator of adenylyl cyclase, on the regulation of CNP-induced pulse pressure in perfused beating rat hypoxic atria. Data were expressed as mean ± SEM, n = 6. *P < 0.05 vs. control group; #P < 0.05 vs. hypoxia group; ♦P < 0.05 vs. CNP group; &P < 0.05 vs. forskolin group.

Figure 3. Effects of CNP (0.1 µmol/L) and forskolin (0.1 µmol/L) on hypoxia- induced atrial phosphodiesterase subtype 3A (PDE 3A) expression. Con, control; Hy, hypoxia; F, forskolin; C, CNP. Data were expressed as mean ± SEM, n = 5. *P < 0.05 vs. control group; #P < 0.05 vs. hypoxia group; ♦P < 0.05 vs. CNP group; &P < 0.05 vs. forskolin group.
3.4. Effects of CNP and Forskolin on HIF-1α Levels in Hypoxic Atria
We next investigated regulation by CNP of hypoxia-induced increases in atrial HIF-1α, as well as the impact of forskolin. As shown in Figure 4, hypoxia substantially increased atrial levels of HIF-1α (P < 0.05 vs. control group) and this effect was completely abolished by CNP (P < 0.05 vs. hypoxia group). In addition, forskolin clearly augmented the hypoxia-induced increase in HIF-1α levels in the atria (P < 0.05 vs. control group; P < 0.05 vs. hypoxia group). This effect was dramatically attenuated by CNP, though HIF-1α levels remained elevated, as compared with hypoxic atria with CNP alone (P < 0.05 vs. control group; P < 0.05 vs. hypoxia group; P < 0.05 vs. CNP group). These results suggested that, in hypoxic atria, CNP suppressed upregulation of HIF-1α and that this effect could be modulated by the AC activator forskolin.
4. Discussion
In our study, in isolated perfused beating rat atria under hypoxic conditions, CNP inhibited atrial dynamics and suppressed HIF-1α levels. This effect of CNP was modulated by the AC activator forskolin, through activation of CNP-PDE 3A signaling.
It is well known that CNP can bind to B-type natriuretic peptide receptors (NPR-B) and negatively affect cardiac myocyte function through activation of the guanylyl cyclase (GC)-cGMP-PKG signaling pathway [17] [18] [19] . In addition, particulate GC (pGC) activation in atria by CNP led to increased pGC- cGMP-PDE 3 signaling and elevated cAMP levels [20] . In our study, hypoxia

Figure 4. Effects of forskolin (0.1 µmol/L) and CNP (0.1 µmol/L) on hypoxia-induced atrial HIF-1α expression. Con, control; Hy, hypoxia; F, forskolin; C, CNP. Data were expressed as mean ± SEM, n = 5. *P < 0.05 vs. control group; #P < 0.05 vs. hypoxia group; ♦P < 0.05 vs. CNP group; &P < 0.05 vs. forskolin group.
significantly inhibited atrial dynamics, an effect clearly augmented by CNP treatment. Furthermore, under hypoxia, the AC activator forskolin substantially increased atrial PDE 3A protein levels without affecting dynamics. Nevertheless, under hypoxia and in the presence of forskolin, the inhibitory effect of CNP on atrial dynamics was substantially attenuated, with concomitant suppression of the forskolin-induced increases in PDE 3A protein levels. These results indicated that CNP can have negative inotropic effects on atrial dynamics under hypoxia and that these actions can be modulated by forskolin, through activation of CNP- PDE 3A signaling. Our results agreed well with previous findings [17] [18] [19] [20] .
Intracellular cAMP is levels are determined by the rate of cAMP generation, through activation of adenylyl cyclase, and its degradation by phosphodiesterases (PDEs) [21] . At least four families of PDEs, PDE 1, PDE 2, PDE 3 and PDE 4, were identified in the heart [22] [23] [24] . PDE 3, a cGMP-inhibited PDE subtype, represents one of the major cAMP-degrading PDEs in the human heart [25] . PKA enhances HIF-1α transcriptional activity [7] [8] and PDE 3 inhibition leading to an elevating of intracellular cAMP levels [20] , which subsequently, activates PKA. Thus, PDE 3 may be involved in regulation of HIF-1α activity. In our study, hypoxia significantly increased atrial HIF-1α protein levels and this effect was augmented by forskolin, which also increased PDE 3A levels. The effect was, in contrast, blocked by CNP, without affecting PDE 3A levels. Nevertheless, the inhibitory effect of CNP on hypoxia-induced atrial HIF-1α protein expression was dramatically attenuated by forskolin. Under these conditions, there was concomitant suppression, by CNP, of the forskolin-induced increases in PDE 3A levels. These results indicated that the suppression by CNP of hypoxia-induced HIF-1α elevation in the perfused beating rat atria was modulated by forskolin, through activation of CNP-PDE 3 signaling. Thus, PDE 3 is a potential regulatory target for modulating HIF-1α activity.
5. Conclusion
In conclusion, CNP inhibited atrial dynamics and HIF-1α activity in the isolated perfused beating rat atria under hypoxic conditions. These effects of CNP were modulated by the AC activator, forskolin, through increased CNP-PDE 3A signaling.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81360061 and 81660089) and a of Jilin Province educational project (No. 2015-44).
Cite this paper
Guan, C.M., Jia, Y.N., Bian, C.C., Zhang, B., Ding, D.Z. and Cui, X. (2017) Forskolin Modulates the Inhibitory Effect of C-Type Natriuretic Peptide on Hypoxia-Induced Atrial Dynamics and Hypoxia Inducible Factor 1 Alpha Activity. Journal of Biosciences and Medicines, 5, 1-10. http://dx.doi.org/10.4236/jbm.2017.51001
References
- 1. van der Pouw Kraan, T.C., Bernink, F.J., Yildirim, C., Koolwijk, P., Baggen, J.M., Timmers, L., Beek, A.M., Diamant, M., Chen, W.J., van Rossum, A.C., van Royen, N., Horrevoets, A.J. and Appelman, Y.E. (2014) Systemic Toll-Like Receptor and Interleukin-18 Pathway Activation in Patients with Acute ST Elevation Myocardial Infarction. Journal of Molecular and Cellular Cardiology, 67, 94-102.
https://doi.org/10.1016/j.yjmcc.2013.12.021 - 2. Mirtschink, P. and Krek, W. (2016) Hypoxia-Driven Glycolytic and Fructolytic Metabolic Programs: Pivotal to Hypertrophic Heart Disease. Biochimica et Biophysica Acta, 1863, 1822-1828.
https://doi.org/10.1016/j.bbamcr.2016.02.011 - 3. Guo, Q., Xu, H., Yang, X., Zhao, D., Liu, S., Sun, X. and Huang, J.A. (2016) Notch Activation of Ca2+-Sensing Receptor Mediates Hypoxia-Induced Pulmonary Hypertension. Hypertension Research.
https://doi.org/10.1038/hr.2016.118 - 4. Mottet, D., Ruys, S.P., Demazy, C., Raes, M. and Michiels, C. (2005) Role for Casein Kinase 2 in the Regulation of HIF-1 Activity. International Journal of Cancer, 117, 764-774.
https://doi.org/10.1002/ijc.21268 - 5. Semenza, G.L. (2012) Hypoxia-Inducible Factors in Physiology and Medicine. Cell, 148, 399-408.
https://doi.org/10.1016/j.cell.2012.01.021 - 6. Eckle, T., Köhler, D., Lehmann, R., El Kasmi, K. and Eltzschig, H.K. (2008) Hypoxia-Inducible Factor-1 Is Central to Cardioprotection: A New Paradigm for Ischemic Preconditioning. Circulation, 118, 166-175.
https://doi.org/10.1161/CIRCULATIONAHA.107.758516 - 7. Bullen, J.W., Tchernyshyov, I., Holewinski, R.J., DeVine, L., Wu, F., Venkatraman, V., Kass, D.L., Cole, R.N., Van Eyk, J. and Semenza, G.L. (2016) Protein Kinase A-Dependent Phosphorylation Stimulates the Transcriptional Activity of Hypoxia-Inducible Factor 1. Science Signaling, 9, ra56.
https://doi.org/10.1126/scisignal.aaf0583 - 8. McNamee, E.N., Vohwinkel, C. and Eltzschig, H.K. (2016) Hydroxylation-Independent HIF-1α Stabilization through PKA: A New Paradigm for Hypoxia Signaling. Science Signaling, 9, fs11.
https://doi.org/10.1126/scisignal.aaf4630 - 9. Zhang, Q.L., Cui, B.R., Li, H.Y., Li, P., Hong, L., Liu, L.P., Ding, D.Z. and Cui, X. (2013) MAPK and PI3K Pathways Regulate Hypoxia-Induced Atrial Natriuretic Peptide Secretion by Controlling HIF-1 Alpha Expression in Beating Rabbit Atria. Biochemical and Biophysical Research Communications, 438, 507-512.
https://doi.org/10.1016/j.bbrc.2013.07.106 - 10. Anttila, K., Streng, T., Pispa, J., Vainio, M. and Nikinmaa, M. (2016) Hypoxia Exposure and B-Type Natriuretic Peptide Release from Langendorff Heart of Rats. Acta Physiologica.
https://doi.org/10.1111/apha.12767 - 11. Hong, L., Xi, J., Zhang, Y., Tian, W., Xu, J., Cui, X. and Xu, Z. (2012) Atrial Natriuretic Peptide Prevents the Mitochondrial Permeability Transition Pore Opening by Inactivating Glycogen Synthase Kinase 3β via PKG and PI3K in Cardiac H9c2 Cells. European Journal of Pharmacology, 695, 13-19.
https://doi.org/10.1016/j.ejphar.2012.07.053 - 12. Hopkins, W.E., Chen, Z., Fukagawa, N.K., Hall, C., Knot, H.J. and LeWinter, M.M. (2004) Increased Atrial and Brain Natriuretic Peptides in Adults with Cyanotic Congenital Heart Disease: Enhanced Understanding of the Relationship between Hypoxia and Natriuetic Peptide Secretion. Circulation, 109, 2872-2877.
https://doi.org/10.1161/01.CIR.0000129305.25115.80 - 13. Rosón, M.I., Toblli, J.E., Della Penna, S.L., Gorzalczany, S., Pandolfo, M., Cavallero, S. and Fernández, B.E. (2006) Renal Protective Role of Atrial Natriuretic Peptide in Acute Sodium Overload-Induced Inflammatory Response. American Journal of Nephrology, 26, 590-601.
https://doi.org/10.1159/000098148 - 14. Cao, X., Xia, H.Y., Zhang, T., Qi, L.C., Zhang, B.Y., Cui, R., Chen, X., Zhao, Y.R. and Li, X.Q. (2015) Protective Effect of Lyophilized Recombinant Human Brain Natriuretic Peptide on Renal Ischemia/Reperfusion Injury in Mice. Genetics and Molecular Research, 14, 13300-13311.
https://doi.org/10.4238/2015.October.26.26 - 15. Liu, L.P., Hong, L., Yu, L., Li, H.Y., Ding, D.Z., Jin, S.J. and Cui, X. (2012) Ouabain Stimulates Atrial Natriuretic Peptide Secretion via the Endothelin-1/ET(B) Receptor-Mediated Pathway in Beating Rabbit Atria. Life Sciences, 90, 793-798.
https://doi.org/10.1016/j.lfs.2012.04.008 - 16. Bian, C., Ding, D., Jin, H., Liu, L., Hong, L., Cui, B. and Cui, X. (2016) EndogenousEndothelin-1 Regulates Hypoxia-Induced Atrial Natriuretic Peptide Secretion by Activating the MAPK/ERK and PI3K/Akt Signaling Pathways in Isolated Beating Rabbit Atria. Journal of Biosciences & Medicines, 4, 45-53.
https://doi.org/10.4236/jbm.2016.41006 - 17. Tan, T., Scholz, P.M. and Weiss, H.R. (2010) Hypoxia Inducible Factor-1 Improves the Negative Functional Effects of Natriuretic Peptide and Nitric Oxide Signaling in Hypertrophic Cardiac Myocytes. Life Sciences, 87, 9-16.
https://doi.org/10.1016/j.lfs.2010.05.002 - 18. Lee, S.J., Kim, S.Z., Cui, X., Kim, S.H., Lee, K.S., Cheng, Y.J. and Cho, K.W. (2000) C-Type Natriuretic Peptide Inhibits ANP Secretion and Atrial Dynamics in Perfused Atria: NPR-B-cGMP Signaling. American Journal of Physiology—Heart and Circulatory Physiology, 278, H208-H221.
- 19. Ding, D.Z., Cui, X., Jin, X.N., Lan, Y., Liu, L.P. and Hong, L. (2010) Effect of C-Type Natriuretic Peptide on Atrial Dynamics in Beating Rabbit Atria and Mechanism of Action. Journal of Jilin University, 36, 23-26.
- 20. Wen, J.F., Cui, X., Jin, J.Y., Kim, S.M., Kim, S.Z., Kim, S.H., Lee, H. and Cho, K.W. (2004) High and Low Gain Switches for Regulation of cAMP Efflux Concentration: Distinct Roles for Particulate GC- and Soluble GC-cGMP-PDE3 Signaling in Rabbit Atria. Circulation Research, 94, 936-943.
https://doi.org/10.1161/01.RES.0000123826.70125.4D - 21. Cui, X., Wen, J.F., Jin, H., Li, D., Jin, J.Y., Kim, S.H., Kim, S.Z., Lee, H.S. and Cho, K.W. (2002) Subtype-Specific Roles of cAMP Phosphodiesterases in Regulation of Atrial Natriuretic Peptide Release. European Journal of Pharmacology, 451, 295- 302.
https://doi.org/10.1016/S0014-2999(02)02294-X - 22. Shahid, M. and Nicholson, C.D. (1990) Comparison of Cyclic Nucleotide Phosphodiesterase Isozymes in Rat and Rabbit Ventricular Myocardium: Positive Inotropic and Phosphodiesterase Inhibitory Effects of Org 30029, Milrinone and Rolipram. Naunyn-Schmiedeberg’s Archives of Pharmacology, 342, 698-705.
https://doi.org/10.1007/BF00175715 - 23. Fischmeister, R. and Hartzell, H.C. (1991) Cyclic AMP Phosphodiesterases and Ca2+ Current Regulation in Cardiac Cells. Life Sciences, 48, 2365-2376.
https://doi.org/10.1016/0024-3205(91)90369-M - 24. Beavo, J.A. (1995) Cyclic Nucleotide Phosphodiesterases: Functional Implications of Multiple Isoforms. Physiological Reviews, 75, 725-748.
- 25. Lakics, V., Karran, E.H. and Boess, F.G. (2010) Quantitative Comparison of Phosphodiesterase mRNA Distribution in Human Brain and Peripheral Tissues. Neuropharmacology, 59, 367-374.
https://doi.org/10.1016/j.neuropharm.2010.05.004