Open Journal of Endocrine and Metabolic Diseases
Vol.4 No.8(2014), Article
ID:48764,8
pages
DOI:10.4236/ojemd.2014.48020
Mitochondrial Dysfunction in Duchenne Muscular Dystrophy
Marie Kelly-Worden, Emily Thomas
Ball State University, Muncie, USA
Email: mlkellyworde@bsu.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 April 2014; revised 20 May 2014; accepted 28 June 2014
ABSTRACT
Muscular Dystrophy (MD) is an X-linked recessive disease affecting mainly boys at a rate of 1 in every 3500 live births. The most common and severe form of the disease is Duchenne Muscular Dystrophy (DMD). The disease is characterized by a relatively rapid wasting of skeletal muscle tissue to a point that leads to paralysis in all patients that suffer from the disease. Unfortunately, due to respiratory or cardiac muscle failure, death occurs in most patients around the age of 30. Currently, the lack of the protein dystrophin is thought to be the chief cause of disease in DMD patients. In addition to a lack of dystrophin, studies are emerging that are painting a picture of a more intricate connection between mitochondrial dysfunction and DMD where increased intracellular and inter-mitochondrial calcium has been shown to cause mitochondrial swelling, loss of mitochondrial membrane integrity, cell death and muscle atrophy. In this article, we will discuss the evidence that places the mitochondrion as a central participant in the etiology of DMD and describe how the relationship between increased intracellular calcium, mitochondrial permeability and dysfunction culminates in muscle loss.
Keywords:Duchene Muscular Dystrophy, Mitochondria, Calcium Homeostasis, Oxidative Stress

1. Introduction
Muscular Dystrophy (MD) is a progressive degenerative disease characterized by slow atrophy of skeletal and cardiac muscle occurring most prominently in boys. Symptoms of the disease usually appear 3 - 5 years after birth, and patients are usually wheelchair bound by the age of 11 - 12 [1] -[3] . Those afflicted with MD usually die before the age of 30 due to cardiac myopathy and/or respiratory failure [1] [2] . The gene behind MD is an X-linked recessive gene and is present in 1 out of every 3500 live births [1] [3] .
Several types of MD exist which vary in both severity and prevalence of the disease. Duchenne (DMD) and Becker Muscular Dystrophy (BMD) are two of the most common and severe forms of MD [1] . The latter is far less severe than the former. DMD and BMD develop as a result of mutations to the dystrophin gene, located on the p arm of the X chromosome [2] . Although a multitude of mutations exist, with about 1 in 3 cases resulting in a novel mutation, the disease is always characterized by reduced or complete absence of dystrophin [1] .
Dystrophin is a protein responsible for myofibril contraction and membrane stability. Since it is lacking or misfolded in DMD and BMD, respectively, myofibril contraction is impaired, and membrane stability is significantly decreased [4] . Muscle membrane fragility and calcium dysregulation go hand in hand in dystrophin deficient muscular dystrophy [5] .
Ca2+ dysregulation has been implicated in MD as well as defective apoptotic pathways and is associated with several classes of diseases, including cancer, autoimmune diseases, and neurodegenerative diseases [6] [7] . As previously mentioned, DMD is characterized by a deficiency of the dystrophin protein which is believed to eventually lead to increased Ca2+ permeability into muscle cells [4] . Because mitochondria can act as a low affinity, high capacity Ca2+ buffer system, the increased permeability of the cell to Ca2+ promotes Ca2+ overload of the mitochondria. This causes the mitochondria to swell [1] [2] [4] , and eventually lose its function. The pathophysiological increase of Ca2+ can induce activation of apoptotic pathways leading to cell death and necrosis (Figure 1).
2. Literature Review
2.1. The Role of Dystrophin and the Hypothesis of Ca2+ Overload
The lack of the dystrophin protein has been found to play a major part in the pathology of DMD and BMD [1] [2] . In muscle biopsies of human and animal muscle tissue, elevated Ca2+ is present, but the mechanisms causing increased Ca2+ are still being determined. Currently, it is believed that a lack of dystrophin causes micro-tears in the sarcolemma due to membrane fragility [2] [4] . This causes abnormal Ca2+ entry and mitochondrial Ca2+ overload. The overload triggers the mitochondrial permeability transition (MPT) mediated by the formation of a large pore protein that spans both inner and outer mitochondrial membranes [2] . Prolonged MPT causes the mitochondrion to lose its inner mitochondrial membrane and matrix leading to swelling and eventual death of the organelle as well as the cell, resulting in the muscle atrophy observed in DMD [2] .
2.2. Biochemical Changes
The exact cause of the biochemical and metabolic changes that lead to muscle atrophy have yet to be determined, but several theories exist that hypothesize the cause of the metabolic changes. In healthy tissue, mitochondria are responsible for ATP production. Reactive oxygen species (ROS) production results as a consequence of ATP production, but is limited in healthy mitochondria. Mitochondria that overproduce radicals lead to cell death via the initiation of the apoptotic pathway [6] -[8] .
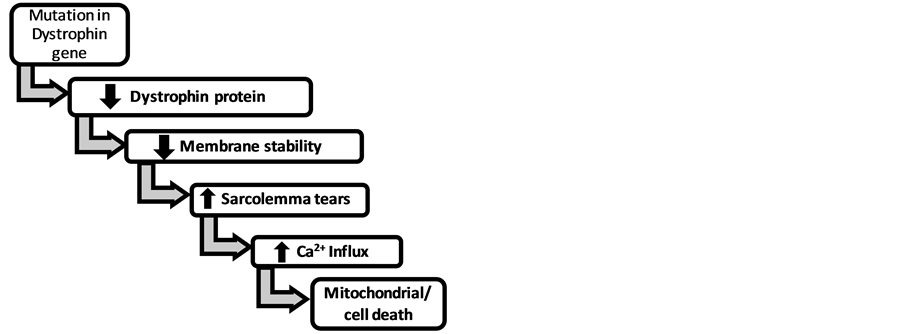
Figure 1. Proposed mechanism for cell death in dystrophin deficient muscular dystrophy.
The primary process within the cell used to synthesize large amounts of ATP is oxidative phosphorylation (ox phos). This process involves multiple inner mitochondrial membrane bound proteins. Several of these proteins are closely regulated by Ca2+ [9] .
The role Ca2+ plays in mitochondrial and eventual ox phos dysfunction is multidimensional. Mitochondrialcalcium sequestration supports at least three physiological processes: 1) to stimulate and control the rate of oxidative phosphorylation; 2) to induce MPT and apoptotic cell death; and 3) to modify the shape of cytosolic calcium pulses or transients [10] -[14] .
Ca2+ has been shown to upregulate the activity of several metabolic enzymes involved in the Krebs cycle including pyruvate dehydrogenase (PDH), alpha-ketoglutarate dehydrogenase (αKDH), and glycerol-3-phospate dehydrogenase (G3PDH) at normal physiological concentrations [9] [15] . In contrast, these vital Krebs cycle enzymes show decreased activity under conditions of Ca2+ overload.
The decrease in activity of these vital Krebs cycle proteins would be expected to result in a decrease in glucose metabolism. Consistent with this idea, Sharma and colleagues demonstrated a significant decrease in glucose metabolism in patients with DMD [16] . In addition, they demonstrated reduced concentrations of several gluconeogenic intermediates in DMD muscle tissue in conjunction with high levels of glycolytic, Krebs cycle, and β-oxidation intermediates suggesting that not only does the cell have trouble metabolizing glucose, but this trouble occurs at multiple levels [16] [17] . Overall, the cell has insufficient means to produce adequate amounts of ATP leading to necrosis of the muscle tissue.
In dystrophin deficient muscle, the influx of Ca2+ causes dysfunction of several ox phox enzyme systems. For example, Ca2+ modulates the activity of enzyme systems including Complex III and the cytochrome chain [10] . Interestingly, these increases are accompanied by an observed decrease in Complex V ATP synthase activity [10] . Perhaps these detected changes account in part, for both the increased production of reactive oxygen species as well as a decrease in ATP production in DMD muscle.
2.3. Oxidative Stress in Dystrophic Tissue
Free radicals and reactive oxygen species (ROS) are regularly formed in healthy tissues under normal physiological conditions. Several factors can increase the formation of ROS and free radicals including exposure to various forms of radiation, exposure to heavy metals, prolonged, unnecessary exposure to UV rays, etc. [18] . Although these species have a propensity to be damaging, their effects are mitigated through antioxidants [19] and specific organelles in healthy cells. With impairment of mitochondrial function in dystrophic tissue, damaging free radicals are increased. Thus, the cell incurs ever increasing amounts of cellular damage, eliciting apoptotic signaling pathways within the mitochondria, and eventually the entire cell [7] .
Ca2+ has been implicated in increased formation of ROS in several diseases including Lou Garrett’s Disease (ALS), various forms of epilepsy, and more recently DMD [8] [18] . The increase in the ROS, leads to the onset of apoptotic pathways. This may be due to the fact that increased Ca2+ load within the organelle impairs the mitochondria’s ability to reduce free radicals [20] . Mitochondria are also highly susceptible to ROS-induced injury because of the close proximity of the mitochondrial DNA to the electron transport chain and due to the lack of protective histones. The accumulation of ROS impairs mitochondrial metabolism and induces oxidative changes in the proteins. ROS can also lead to the induction of the mitochondrial permeability transition (MPT) [21] .
2.4. The Relationship of MPT, ROS and the Mitochondrial Membrane Potential ((m)
MPT is defined by an increase of mitochondrial inner membrane permeability to ions and solutes with molecular masses of up to about 1500 Da that leads to matrix swelling [22] . MPT is the result of the opening of the mitochondrial megachannel according to the permeability pore theory which was established by Zoratti et al., 2005 [22] -[25] . This theory is supported by the fact that both the megachannel and MPT are sensitive to cyclosporin A. Cyclosporin A inhibits MPT after binding to cyclophilin D, whose enzymatic activity is blocked by cyclosporin A [22] [26] [27] in the same concentration range that inhibits the megachannel [22] [28] [29] .
The cause of increased Ca2+ permeability into myocytes seems well established. However, the cause of increased mitochondrial membrane permeability to Ca2+ remains to be determined. The driving force behind Ca2+ uptake is the mitochondrial membrane potential (Ψm). The Ψm is present throughout the inner mitochondrial membrane and is generated by the respiratory chain. Pumping of protons by the respiratory chain toward the intermembrane space generates the electrochemical gradient inside the mitochondrial matrix. Ca2+ entry would be expected to be promoted by this gradient, however, two strong uncouplers of oxidative phosphorylation exist, dinitrophenol and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) that typically prevent Ca2+ entry. Therefore, mitochondrial Ca2+ uptake occurs via the Ca2+ uniporter and export of Ca2+ from the mitochondrial matrix occurs via a Ca2+ antiporter [30] .
Onopiuk and associates demonstrated several changes to the mitochondria in dystrophic myoblasts [8] . This study is of particular interest due to the lack of dystrophin in both diseased and healthy cells. Upon observation of the cells, Onopiuk et al., 2009, noticed that diseased cells demonstrated several changes associated with mature muscle tissue. The changes observed included increased membrane potential in the outer mitochondrial membrane, decreased oxygen consumption and increased production of ROS.
Mitochondria are the chief source of ROS formation in the cell. At low concentrations of calcium, the MPT pore when activated results in a decrease in ROS production due to partial mitochondrial depolarization. At high calcium concentrations, MPT pore activation results in an increase of ROS release from the mitochondria due to the rapid transition to the non-equilibrium state because of cytochrome c loss and progressive gating of electron flow via the electron transport chain [31] .
The transition to the non-equilibrium state, in response to the release of cytochrome c and progressive respiration inhibition, results in the loss of proportionality in the rate of ROS production to the rate of respiration and subsequently a rise in ROS production over time. This rise in ROS production is stated to be independent of membrane potential. This is not exactly true since the rate of ROS formation is controlled by the rate of membrane potential dependent Ca2+ uptake which is the rate-limiting step in ROS production [31] .
Indeed, Skulachev, 1996, and Korshunov et al., 1997, found ROS generation to be strongly dependent on mitochondrial membrane potential (Ym) and believed that the loss of cytochrome c was a last ditch effort of the mitochondria before apoptosis. They found that there existed a threshold value for Ym above which even a small increase in Ym gave rise to a large stimulation of H2O2 production by the mitochondria [32] [33] .
Either way, chronic increases in ROS formation are responsible for the accumulation of ROS-associated damages. These damages include damage to DNA, proteins, and lipids. The resulting damage leads to progressive cell dysfunctions and eventually apoptosis ([34] , Figure 2).
2.5. The Hypothesis of Mitochondrial Dysfunction in DMD
In Onopiuk’s study they also observed a decreased presence of mitochondrial complexes I, III, IV and V (ATP synthase) as well as ATP production [6] [8] . ATP production was not altered in dystrophic cells treated with
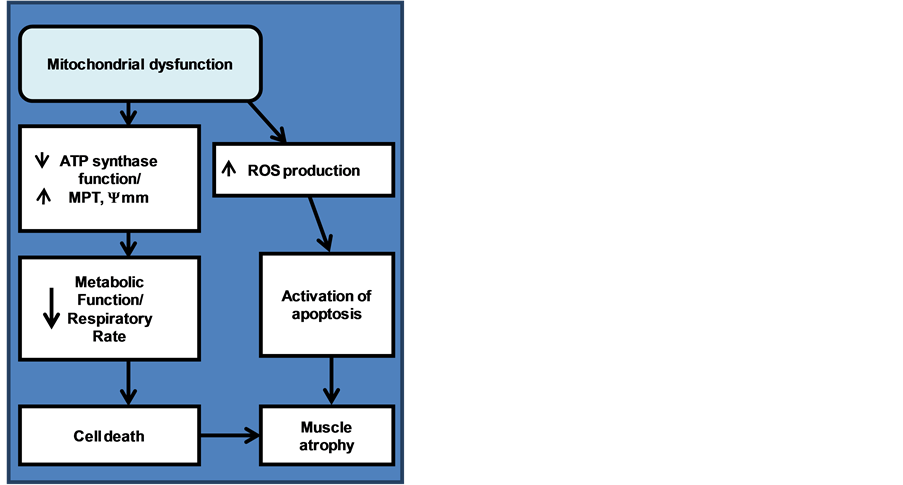
Figure 2. Development of the pathway from mitochondrial dysfunction to muscle atrophy.
oligomycin which decreases ATP synthase activity in healthy cells [8] . This result supports a role for ATP synthase dysfunction in dystrophic tissue.
The ATP synthase is formed by the catalytic unit F1, the membrane-bound proton-translocating unit F0, and the lateral stalk that links F1 and F0 [22] [35] -[39] . Cyclophilin D attaches to this stalk. The F0F1 ATP synthase complex associates to form dimers and the dimers are hypothesized to represent the physiological unit of Complex V in the mitochondrial inner membrane [22] [36] .
F0F1 dimers when incorporated into azolectin bilayers present a conductance with characteristics that closely match those of the megachannel and MPT [40] . Therefore, the megachannel, MPT, and F0F1 dimers are believed to be one in the same [22] . Even more convincing to this point, was a study done by Csukly et al., 2006, where it was discovered that muscle denervation promoted MPT and increased the expression of cyclophilin D [41] . Increased cyclophilin D resulted in an increased sensitivity of calcium induced activation of the megachannel/MPT. Likewise, treatment of the DMD mouse model with Debio 025, a cyclophilin inhibitor, resulted in partial improvement of the structure and the function of the dystrophic mouse muscle [42] . Thus, one can conclude that mitochondrial dysfunction and the MPT play a role in DMD. More specifically, this role is associated with a decrease in ATP synthase function and an increase in the megachannel/MPT.
To further support this argument, Pellegrini and colleagues in 2013, found that melanocytes taken from patients with DMD possessed mitochondria that readily accumulated tetramethylrhodamine. The uptake of this compound implies that the mitochondria are energized irrespective of the presence of dystrophin. Both the melanocytes and myoblasts of DMD patients hyperpolarized as would be expected upon addition of the F0F1 ATPase inhibitor oligomycin, however, this was followed by a rapid depolarization. This suggests that the cells are affected by a latent mitochondrial dysfunction that is unmasked by inhibition of the ATP synthase [43] .
Since these changes in mitochondrial function can be observed even before dystrophic cells express dystrophin, changes in mitochondrial function occur before muscle membrane breakdown and therefore, precede the theory of Ca2+ overload. Therefore, it very well maybe defective mitochondria that lead to energetic deficiency and play a role in promoting myofibril atrophy via the opening of MPT [44] .
If we begin with the perception that mitochondrial dysfunction may exist even before Ca2+ overload, we need to take into account the role of mitochondria as the main producers of ATP. ATP is necessary to provide the energy for pumps within the cell including those needed for Ca2+ homeostasis. The dysfunctioning mitochondria possess a decrease in proteins related to ATP production including the ATP synthase. The result is a decrease in ATP production. The decrease in ATP production is coupled with an increase in Ym. A high protonic potential actuates production of ROS in the mitochondria [32] [33] . Generation of ROS by the mitochondria leads to further mitochondrial dysfunction.
It has been suggested that differences exist in pre-symptomatic and post-symptomatic patients with DMD. Pre-symptomatic patients were found to have a down regulation in the genes encoding the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) in cardiac muscle and an increase in PKA-related genes. This overexpression of PKA-related genes along with a decrease in the pholamban (PLN) to SERCA ratio is thought to promote the flux of calcium from the cytoplasm into the endoplasmic reticulum [45] .
However, with time and age, the decrease in mitochondrial function and resulting deduction in ATP leads to a decrease in the energy to power SERCA. The result is a buildup of Ca2+ in the cytosol. This is perhaps where the boundaries between Ca2+ overload and mitochondrial dysfunction cross. In this case, the mitochondria are then not just the victims of Ca2+ overload, but active participants in calcium dysregulation. This hypothesis will need to be addressed in research related to DMD and should be looked at for other forms of multiple dystrophy as well.
3. Conclusions
The concept of mitochondrial dysfunction in DMD is well supported by the findings of Onopiuk. The increase in membrane potential, the decrease in respiratory rate and the decrease in mitochondrial complexes all promote mitochondrial dysfunction. Although Ca2+ dysregulation may play a role in ROS generation, changes in Ym that lead to ROS generation and induction of impairment in mitochondria more strongly support DMD properties than Ca2+ overload. The extremely high-maximal potential that can be generated may be the result of loss of activity associated with the decrease of respiratory rate and ATP synthase function.
Overall, it is the loss of mitochondrial activity that is also the cause of sarcopenia with senescence observed in DMD patients. Therefore, mitochondrial dysfunction is a very probable mechanism for the pathology of DMD and may act as a contributor along with dystrophin impaired membranes to promote the observed Ca2+ overload. The combination of mitochondrial dysfunction, ROS production and ATP depletion along with the ever increasing calcium accumulation lead to cell death and muscle atrophy.
Acknowledgements
We would like to acknowledge DR Giovannucci for editorial commentary on this paper.
References
- Honeycomb, J. and Anand, R. (1989) Molecular Analysis of the Duchenne Muscular Dystrophy Locus.
Comparative Biochemistry and Physiology, 93A, 125-131. http://dx.doi.org/10.1016/0300-9629(89)
90199-0 - Millay, D.P., Sargent, M.A., Osinska, H., Baines, C.P., Barton, E.R., Vuagniaux, G., Sweeney, H.L., Ro-
bbins, J. and Molkentin, J.D. (2008) Genetic and Pharmacologic Inhibition of Mitochondrial Dependent
Necrosis Attenuates Muscular Dystrophy. Nature Medicine, 14, 442-447. http://dx.doi.org/10.1038/
nm1736 - Perumal, A.R., Rajeswaran, J. and Nalini, A. (2013) Neuropsychological Profile of Duchenne Muscular Dystrophy. Applied Neuropsychology: Child. http://dx.doi.org/10.1080/21622965.2013.802649
- Deconinck, N. and Dan, B. (2007) Pathophysiology of Duchenne Muscular Dystrophy: Current Hypotheses. Pediatric Neurology, 36, 1-7. http://dx.doi.org/10.1016/j.pediatrneurol.2006.09.016
- Jorgensen, L.H., Blain, A., Greally, E., Laval, S.H., Blamire, A.M., Davison, B.J., Brinkmeier, H., MacGowan, G.A., Schroder, H.D., Bushby, K., Straub, V. and Lochmüller, H. (2011) Long-Term Blocking of Calcium Channels in mdx Mice Results in Differential Effects on Heart and Skeletal Muscle. American Journal of Pathology, 178, 273-283. http://dx.doi.org/10.1016/j.ajpath.2010.11.027
- Albrekkan, F.M. and Kelly-Worden, M. (2013) Mitochondrial Dysfunction and Alzheimer’s Disease. Op-
en Journal of Endocrine and Metabolic Diseases, 3, 14-19. http://dx.doi.org/10.4236/ojemd.2013.32
A003 - Pinton, P., Giorgi, C., Siviero, R., Zecchini, E. and Rizzuto, R. (2008) Calcium and Apoptosis: ER-Mitoc-
hondrial Ca2+ Transfer and the Control of Apoptosis. Oncogene, 27, 6407-6418. http://dx.doi.org/1
0.1038/onc.2008.308 - Onopiuk, M., Brutkowski, W., Wierzbika, K., Wojcichowska, S., Szczepanowka, J., Fronk, J., Lochmuller, H., Gorecki, D.C. and Zablocki, K. (2009) Mutation in Dystrophin-Encoding Gene Affects Energy Metabolism in Mouse Myoblasts. Biochemical and Biophysical Research Communications, 386, 463-466. http://dx.doi.org/10.1016/j.bbrc.2009.06.053
- Kavanagh, N., Ainscow, E.K. and Brand, M.D. (2000) Calcium Regulation of Oxidative Phosphorylation
in Rat Skeletal Muscle Mitochondria. Biochemica et Biophysica Acta, 1457, 57-70. http://dx.doi.org/10.
1016/S0005-2728(00)00054-2 - Gunter, T.E., Yule, D.I., Gunter, K.K., Eliseev, R.A. and Salter, J.D. (2004) Calcium and Mitochondria. FASEB Letters, 567, 96-102. http://dx.doi.org/10.1016/j.febslet.2004.03.071
- Godin, R., Daussin, F., Matecki, S., Li, T., Petrof, B.J. and Burelle, Y. (2012) Peroxisome Proliferator-Activated Receptor γ Coactivator1-α Gene Transfer Restores Mitochondrial Biomass and Improves Mitochondrial Calcium Handling in Post-Necrotic mdx Mouse Skeletal Muscle. The Journal of Physiology, 590, 5487-5502. http://dx.doi.org/10.1113/jphysiol.2012.240390
- Green, D.R. and Kroemer, G. (2004) The Pathophysiology of Mitochondrial Cell Death. Science, 305, 626-629. http://dx.doi.org/10.1126/science.1099320
- Zamzami, N., Larochette, N. and Kroemer, G. (2005) Mitochondrial Permeability Transition in Apoptosis and Necrosis. Cell Death and Differentiation, 12, 1478-1480.
http://dx.doi.org/10.1038/sj.cdd.4401682 - Burelle, Y., Khairallah, M., Ascah, A., Allen, B.G., Deschepper, C.F., Petrof, B.J. and Rosiers, D.C. (2010) Alterations in Mitochondrial Function as a Harbinger of Cardiomyopathy: Lessons from the Dystrophic
Heart. Journal of Molecular and Cellular Cardiology, 48, 310-321. http://dx.doi.org/10.1016/j.yjmcc.20
09.09.004 - Glancy, B. and Balaban, R.S. (2012) Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetic. Biochemistry, 51, 2959-2973. http://dx.doi.org/10.1021/bi2018909
- Sharma, U., Atri, S., Sharma, M.C., Sakar, C. and Jagannathan, N.R. (2003) Skeletal Muscle Metabolism in Duchenne Muscular Dystrophy (DMD): An In-Vitro Proton NMR Spectroscopy Study. Magnetic Resonance Imaging, 21, 145-153. http://dx.doi.org/10.1016/S0730-725X(02)00646-X
- Griffin, J.L., Williams, H.J., Sang, E., Clarke, K., Rae, C. and Nicholson, J.K. (2001) Metabolic Profiling of Genetic Disorders: A Multitissue 1H-Nuclear Magnetic Resonance Spectroscopic and Pattern Recogni-
tion Study into Dystrophic Tissue. Analytical Biochemistry, 293, 16-21. http://dx.doi.org/10.1006/abio.
2001.5096 - Naziroglu, M. (2009) Role of Selenium on Calcium Signaling and Oxidative Stress-Induced Molecular Pathways in Epilepsy. Neurochemical Research, 34, 2181-2191. http://dx.doi.org/10.1007/s11064-009-0015-8
- Ma, Q. (2014) Advances in Mechanisms of Anti-Oxidation. Discovery Medicine, 17, 121-130.
- Carriedo, S.G., Sensi, S.L., Yin, H.Z. and Weiss, J.H. (2000) AMPA Exposures Induce Mitochondrial Ca2+ Overload and ROS Generation in Spinal Motor Neurons in Vitro. The Journal of Neuroscience, 20, 240-250.
- Pervaiz, S., Taneja, R. and Ghaffari, S. (2009) Oxidative Stress Regulation of Stem and Progenitor Cells. Antioxidants & Redox Signaling, 11, 2777-2789. http://dx.doi.org/10.1089/ars.2009.2804
- Bernardi, P. (2013) The Mitochondrial Permeability Transition Pore: A Mystery Solved? Frontiers in Physiology, 4, 95. http://dx.doi.org/10.3389/fphys.2013.00095
- Kinnally, K.W., Campo, M.L. and Tedeschi, H. (1989) Mitochondrial Channel Activity Studied by Patch-
Clamping Mitoplast. Journal of Bioenergetics and Biomembranes, 21, 497-506. http://dx.doi.org/10.1
007/BF00762521 - Petronilli, V., Szabó, I. and Zoratti, M. (1989) The Inner Mitochondrial Membrane Contains Ion-Condu-
cting Channels Similar to Those Found in Bacteria. FEBS Letters, 259, 137-143. http://dx.doi.org/10.1
016/0014-5793(89)81513-3 - Zoratti, M., Szabó, I. and De Marchi, U. (2005) Mitochondrial Permeability Transitions: How Many Do-
ors to the House? Biochimica et Biophysica Acta (BBA), Bioenergetics, 1706, 40-52. http://dx.doi.org/
10.1016/j.bbabio.2004.10.006 - Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T. and Schmid, F.X. (1989) Cyclophilin and Peptidyl-Prolyl cis-Trans Isomerase Are Probably Identical Proteins. Nature, 337, 476-478. http://dx.doi.org/10.1038/337476a0
- Takahashi, N., Hayano, T. and Suzuki, M. (1989) Peptidyl-Prolyl cis-Trans Isomerase Is the Cyclosporin A-Binding Protein Cyclophilin. Nature, 337, 473-475. http://dx.doi.org/10.1038/337473a0
- Connern, C.P. and Halestrap, A.P. (1992) Purification and N-Terminal Sequencing of Peptidyl-Prolyl cis-Trans Isomerase from Rat Liver Mitochondrial Matrix Reveals the Existence of a Distinct Mitochondrial Cyclophilin. Biochemical Journal, 284, 381-385.
- Connern, C.P. and Halestrap, A.P. (1994) Recruitment of Mitochondrial Cyclophilin to the Mitochondrial Inner Membrane under Conditions of Oxidative Stress That Enhance the Opening of a Calcium-Sensitive Non-Specific Channel. Biochemical Journal, 302, 321-324.
- Marchi, S. and Pinton, P. (2014) The Mitochondrial Calcium Uniporter Complex: Molecular Components, Structure and Physiopathological Implications. The Journal of Physiology, 592, 829-839. http://dx.doi.org/10.1113/jphysiol.2013.268235
- Akopova, O.V., Kolchynskayia, L.Y., Nosar, V.Y., Smyrnov, A.N., Malisheva, M.K., Man’kovskaia, Y.N. and Sahach, V.F. (2011) The Effect of Permeability Transition Pore Opening on Reactive Oxygen Species Production in Rat Brain Mitochondria. Ukrainskii Biokhimicheskii Zhurnal, 83, 46-55.
- Skulachev, V. (1996) Role of Uncoupled and Non-Coupled Oxidations in Maintenance of Safely Low Levels of Oxygen and Its One-Electron Reductants. Quarterly Reviews of Biophysics, 29, 169-202. http://dx.doi.org/10.1017/S0033583500005795
- Korshunov, S.S., Skulachev, V.P. and Starkov, A.A. (1997) High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Letters, 416, 15-18. http://dx.doi.org/10.1016/S0014-5793(97)01159-9
- Suski, J.M., Lebiedzinska, M., Bonora, M., Pinton, P., Duszynski, J. and Wieckowski, M.R. (2012) Relation between Mitochondrial Membrane Potential and ROS Formation. Methods in Molecular Biology, 810, 183-205. http://dx.doi.org/10.1007/978-1-61779-382-0_12
- Strauss, M., Hofhaus, G., Schroder, R.R. and Kühlbrandt, W. (2008) Dimer Ribbons of ATP Synthase
Shape the Inner Mitochondrial Membrane. EMBO Journal, 27, 1154-1160. http://dx.doi.org/10.1038/
emboj.2008.35 - Thomas, D., Bron, P., Weimann, T., Dautant, A., Giraud, M.F., Paumard, P., et al. (2008) Supramolecular Organization of the Yeast F1F0-ATP Synthase. Biology of the Cell, 100, 591-603. http://dx.doi.org/10.1042/BC20080022
- Rees, D.M., Leslie, A.G. and Walker, J.E. (2009) The Structure of the Membrane Extrinsic Region of Bovine ATP Synthase. Proceedings of the National Academy of Sciences of the United States of America, 106, 21597-21601. http://dx.doi.org/10.1073/pnas.0910365106
- Baker, L.A., Watt, I.N., Runswick, M.J., Walker, J.E. and Rubinstein, J.L. (2012) Arrangement of Subunits in Intact Mammalian Mitochondrial ATP Synthase Determined by cryo-EM. Proceedings of the
National Academy of Sciences of the United States of America, 109, 11675-11680. http://dx.doi.org/
10.1073/pnas.1204935109 - Davies, K.M., Anselmi, C., Wittig, I., Faraldo-Gomez, J.D. and Kühlbrandt, W. (2012) Structure of the Yeast F1F0-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proceedings of the
National Academy of Sciences of the United States of America, 109, 13602-13607. http://dx.doi.org/
10.1073/pnas.1204593109 - Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013) Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proceedings of the National Aca-
demy of Sciences of the United States of America, 110, 5887-5892. http://dx.doi.org/10.1073/pnas.
1217823110 - Csukly, K., Ascah, A., Matas, J., Gardiner, P.F., Fontaine, E. and Burelle, Y. (2006) Muscle Denervation Promotes Opening of the Permeability Transition Pore and Increases the Expression of Cyclophilin D. The Journal of Physiology, 574, 319-327. http://dx.doi.org/10.1113/jphysiol.2006.109702
- Reutenauer, J., Dorchies, O.M., Patthey-Vuadens, O., Vuagniaux, G. and Ruegg, U.T. (2008) Investigation of Debio 025, a Cyclophilin Inhibitor, in the Dystrophic mdx Mouse, a Model for Duchen-
ne Muscular Dystrophy. British Journal of Pharmacology, 155, 574-584. http://dx.doi.org/10.1038/bjp.
2008.285 - Pellegrini, C., Zulian, A., Gualandi, F., Manzati, E., Merlini, L., Michelini, M.E., Benassi, L., Marmiroli, S., Ferlini, A., Sabatelli, P., Bernardi, P. and Maraldi, N.M. (2013) Melanocytes—A Novel Tool to Study Mitochondrial Dysfunction in Duchenne Muscular Dystrophy. Journal of Cellular Physiology, 228, 1323-1331. http://dx.doi.org/10.1002/jcp.24290
- Pauly, M., Daussin, F., Burelle, Y., Li, T., Godin, R., Fauconnier, J., Koechlin-Ramonatxo, C., Hugon, G., Lacampagne, A., Coisy-Quivy, M., Liang, F., Hussain, S., Matecki, S. and Petrof, B.J. (2012) AMPK Activation Stimulates Autophagy and Ameliorates Muscular Dystrophy in the mdx Mouse Diaphragm. American Journal of Pathology, 181, 583-592.
- Tian, L.J., Cao, J.H., Deng, X.Q., Zhang, C.L., Qian, T., Song, X.X. and Huang, B.S. (2014) Gene Expression Profiling of Duchenne Muscular Dystrophy Reveals Characteristics along Disease Progression. Genetics and Molecular Research, 28, 1402-1411.