Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.5A(2013), Article ID:35880,10 pages DOI:10.4236/ojemd.2013.35A001
PPAR and Local Renin-Angiotensin Systems in Cardiovascular and Metabolic Diseases Associated with Obesity: A Unifying Hypothesis
1Center of Integral Attention to People with Diabetes (CAIAD), Corozal, Colombia
2Division of Cardiovascular Sciences, Center for Applied Medical Research and University Clinic, University of Navarra, Pamplona, Spain
Email: piedegus@yahoo.es
Copyright © 2013 Gustavo Márquez-Salom, Javier Diez. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 27, 2013; revised June 27, 2013; accepted July 27, 2013
Keywords: PPAR; Obesity; Diabetes; RAAS; Insulin
ABSTRACT
Recent research has revealed roles for the PPAR family of transcription factors in the modulation of the RAAS. In particular, PPARα and PPARγ have been shown to control the transcription of renin in several tissues and influence the activity of local renin-angiotensin-systems coupling vascular and metabolic functions and contributing to their modulation. In this conceptual framework, PPAR and local RAS participate in the pathophysiology of blood pressure elevation, as well as in organ and tissue damage, and disturbances of glucose and lipid metabolism associated with cardiovascular diseases and obesity. The understanding of these mechanisms here discussed and their adequate pharmacological manipulation enlarge the potential to intervene pathological processes that connect the obesity with associate diseases and could alert about the adverse effects of some PPAR agonists on involved organs.
1. Introduction
The Renin-Angiotensin-Aldosterone System (RAAS) arose when the prehistoric animals began to live outside of the sea and of its high content of sodium and, according to Darwin, only those species that could structure the system, could survive leaving the sea. The maintenance of the arterial pressure and the saving of sodium and of the water when the animal lost blood and got dehydrated or when he wandered for the desert, were necessary while it was able of watering. But also, the energy needs were key points of survival and the RAAS should work coupled with mechanisms of metabolic or energy control, or include them. Therefore, the vision of the RAAS like control system of salt and water and of the vascular tone is short and it should be enlarged until regulatory mechanisms of energy homeostasis, being involved in systemic, tissue and intracellular effects, transcending until conservation, adaptation or cellular apoptosis. PPARs (peroxisome proliferator activated-receptors) are distributed in diverse organs and constitute a complex and integrated mechanism with fundamental functions for the adaptation, modulation and survival of many animal species. PPARs also influence the growth and cellular differentiation, reparative processes, the glucose and lipid metabolism, the thermogenesis, the maintenance of the arterial pressure and salt and water balance, among others. PPARα, γ, β/δ don’t work scatteredly if not that act together to modulate final effects. Its location in the organs and tissues is bound to the same functions of each organ and its corresponding relationship with mechanisms of adaptation, modulation and survival. Maybe without PPAR, the RAAS could not cover the whole spectrum of preservation functions that manages.
Claude Bernard considered that “the physiological processes uncoupling generates pathology and the pathological processes reside in physiologic processes”. Disruptions in PPAR functions have been related with cancer, Alzheimer disease, obesity, diabetes and hypertension among others.
2. PPAR and Local Renin-Angiotensin Systems (RAS)
2.1. PPAR, Visceral Adipocytes and Intra-Adipocytes Renin-Angiotensin System
The differentiation of adipocytes, the modulation of the insulin actions, the capacity to generate inflammation, the contribution to the maintenance of the arterial pressure , the salt and water balance, the reinforcement of the sympathetic adrenal system, they are functions self influenced by the visceral adipocytes which confer to these cells high privileges of important central control where PPAR play regulatory functions over insulin secretion and action, hepatic gluconeogenesis, glucose homeostasis, thermogenesis, lipogenesis, lipolysis and food intake. Many of these actions are bound to the activity of the renin-angiotensin-system (RAS) in these same cells where their hyperactivity could generate hypertension if we take like reference the secretory contribution of angiotensinogen (AGT) for mature adipocytes in animal models and in human, [1-3] which would increase of the angiotensin II (AII) circulating and could be related with activation of the RAS in the visceral adipose tissue. PPARα are involved in the control of the metabolism of glucose and lipids, this way mice PPARα KO in fast or high diet in fatty, develop hypoglycemia, hypoketonemia and high levels of FFA ( free fatty acids) [4,5]. It is also interesting that the absence of PPARα avoids the arterial hypertension and attenuates the atherosclerosis in Tsukuba Hypertensive Mouse (THM) [6.7]. When these animals became THM/PPARαKO, the hypertension and the myocardial hypertrophy were completely abolished, decreased the activity of plasmatic renin and the serum aldosterone was normalized. As well the PPARα binds to hormone responsive element (HRE) in the promoter of the human angiotensin (AGT) gene as PPAR also has been identified in the gene of the renin, suggesting potential regulation at transcriptional level [8,9]. Karlsson and other investigators have demonstrated the presence of renin, angiotensinogen and of the enzymes required for its conversion to angiotensin II (AT II) in the human adipocytes [10]. On the other hand, Harte and collaborators measured the level of circulating AII and demonstrated that it increases in the measure in that the body mass index (BMI) is increased; they have also demonstrated that the increment of tumor necrosis factor α (TNFα) measured in venous drainage of the adipose tissue is made in correlation with the increase of circulating AII and that the delivery of this, stimulated with insulin, decreases notably when adding rosiglitazone, an agonist PPARγ [11], being confirmed this way, the delivery of AII to the circulation from the adipocytes and the effect of the PPARγ agonists to brake this process. We have left the query of if the adipocytes renin here is imported or innate, but today we know that several tissues and organs synthesize it and they have local components of the RAS [12]. Additionally, in human cellular lines expressing renin like CaLu-6, PPARγ acts as a potent and specific activator of the renin gene expression [13]. If this also happens in the adipocytes, the stimulus PPARγ may contribute to the renin expression, at the time that suppresses effects of AII, being structured a local regulator mechanism to modulate the activity of the RAS (Figure 1). Under physiologic conditions, the free fatty acids, natural PPARγ agonists, would stimulate renin activity. There would be a negative feedback to neutralize its expression through the TNFα, which has been studied previously by Todorov [14]. In cases of visceral obesity, the production of FFA increased by the lipolysis, will elicit PPARγ activity, augmenting the renin expression and consecuently AII. In order to avoid RAS over activity, AII would generate signals inducing nuclear factor kappa beta (NF-kβ) and consequently TNFα that would break the expression of the renin gene until certain physiological limits over which it would fail.
In conditions of sustained hyperglycemia and obesity, the physiological homeostasis would fail and the final result could be an increment of the renin and AII actions, which should have consequences in the glucose and lipid metabolism. The visceral adipose tissue has a peptides repertoire, cytokines, and hormones that may group in 2 bands: one, composed by adiponectin, IGF-1 and ghrelin that favors the action of the insulin, and another composed for leptin, resistin, TNFα, IL-6, UCP 2, protein agouti, adipsin, RBP4 and AII that are opposed to the action of the insulin. AII is able to stimulate the activity of the NF-kβ, favoring the production of TNFα, inflammatory citokinas (IL-6, IL-8, IL-1β) and generating oxidative stress. When AII increases, the level of TNFα does, altering the signaling of the insulin in the tissues, modifying the appropriate phosphorilation of the insulin receptor, but also activating protein tirosin phosphatases with the consequent inhibition of the activity of insulin receptor substrate 1 (IRS-1) [15]. While adiponectin favors the appropriate phosphorylation of the insulin in its receptor, AII commits this mechanism [16,17] eliciting signals that induce through Foxo-1/C/EBPα and activation of sirtuin 1 (SIRT1), a transcriptional effect that allows the expression of the adiponectin gene, in a compensatory intent to neutralize the deleterious effect to the
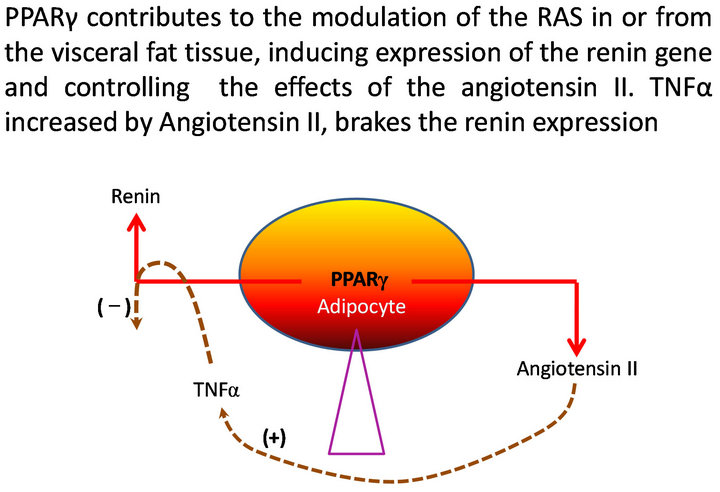
Figure 1. Homeostasis and modulation of the intracellular RAS in visceral adipocytes.
signaling of the insulin caused by AII [18]. Additionally AII supplements its made generating inhibitory signals of the adipocyte differentiation and its maturation in mature adipocytes, which decreases the production of adiponectin that should require well maturated adipocytes. However, Foxo-1, responds in behalf of insulin action, stimulating the factor of growth PDX-1 so that it improves the trophic condition of the pancreatic β cells for producing more insulin [19,20]. Foxo-1 also opposes to those inhibitory differentiation signals over adipocytes maturation originated by AII. PPARγ agonists promote well adipocyte differentiation improving the conditions to secrete adiponectin and counterweight the effects of AII on the insulin actions. We could summarize that PPARγ arise expression of renin and consequently of angiotensin II in the visceral adipocytes, affecting insulin actions, but simultaneously trigger opposite mechanism for improving such effects, establishing a modulator balance. So, these facts show the great importance and interaction of the RAS with the control of the glucose and lipid metabolism influenced by PPARα and PPARγ. The visceral adipose tissue on the other hand and through regulatory functions mediated by PPAR α y γ, influence the control of the vascular tone with vasodilator effect (insulin, adiponectin and ghrelin) sympathetic tone and renal balance of sodium (leptin) and thermogenesis (leptin and UCP-2) [21].
2.2. PPAR, Kidneys and Renin-Angiotensin-Aldosterone System
The studies to investigate the mechanism why do thiazolidinediones (TZD) cause edema and increment weight allowed to know the importance of PPARγ, coded by Pparg in the control of epithelial sodium channels (ENaC) in the collecting ducts [22]. Mice treated with TZD experienced weight gain from increased total body water, which was blocked with amiloride or prevented with deletion of the PParg from the collecting duct, which decreased renal sodium avidity and increased plasma aldosterone. In a same way, PPARα could have important physiological role in helping to maintain the osmotic balance through upward regulation of aquagliceroporin (AQP 3) [23]. In animal models prone to atherosclerosis (diabetic mice apoEKO), both, PPARα (gemfibrozil) and PPARγ (rosiglitazone) agonists showed renal protection in glomerular structure and function [24]. Mild reductions of the arterial blood pressure and microalbuminuria have been also shown in patients receiving TZD. Taking the knowledge about TZD toward the comprehension of their clinical side effects on weight gain and edema, it could be thought that a sustained or strong PPARγ agonism in some individuals may exceed the physiological mechanisms to regulate the body water through ENaC. The aldosterone increment and of the urinary excretion of sodium when deletion of the Pparg is made or amiloride is administered to avoid the PPARγ effect on the ENaC, show a coherent relationship between PPARγ effect and aldosterone inside which both work for the intravascular volume expansion. This way the things, spironolactone was a therapeutic option to diminish the edema caused by thiazolidinediones as well as the addition of and ACE inhibitor to brake the RAAS [25]. PPARγ also brake actions of the AII on the insulin action and its proliferative signals contributing to modulate effects mediated by AII that may intervene in renal injury.
2.3. PPAR, Heart and RAS
PPARγ2 contribute with signals that jointly with members of the C/EBPα family would procure higher differentiation of mesenchymatous cells to adipocytes in detriment of the fibroblasts formation, muscular cells and maker bone cells [26]. This may be an explanation to the osteoporosis and excess of fractures attributable to a sustained PPARγ effect as has met in some clinical trials with TZD [27]. We don’t still know the negative influence that this mechanism may have in the heart in terms of muscular tissue differentiation or growth of fibrous tissue, but we know that very high dose of TZD caused heart hypertrophy in laboratory animals and those hearts of rodents that increased its weight a 25% receiving PPARγ agonists, were associated to a premature heart mortality in 2 years, in studies for carcinogenesis [28]. In the laboratory, high dose of PPARγ agonists resulted in edema, gain of weight, heart hypertrophy and heart failure in all the species, however doses of PPAR agonists with margins of security not associated with higher increment of weight in the heart and not associated with cancer were looked in order to advance to studies phase 2 and 3. Low dose of PPARγ agonists may be, on the contrary, protects against the heart hypertrophy induced in vitro by AII [29,30]. In the heart, as well as in the adipose tissue may happens that PPARγ stimulated by high dose of pharmacological or natural agonists (excess or FFA in obeses) may increase the renin expression, native or imported, through transcriptional way [13,31], without that AII may counterweight it, breaking a physiological balance. In contrast, in a physiological range, PPARγ would induce renin expression at the same time that AII would brake it, establishing a mechanism in which both could act balanced. We should not lose of view that PPARα, γ y β/δ are an integrated system with signals that act synchronized. During the development of the heart hypertrophy, it had been met reduction in the oxidative capacity of de fatty acids and higher utilization of glucose as energetic substrate explainable by reduction in the PPARα effect. PPARαKO mice present reduction in the expression of genes related to the oxidation of fatty acids, deposit fat in their hearts with fibrosis [32]. In humans PPARα could be related with the regulation of the left ventricle growth in response to the exercise and the hypertension [33]. Etomoxir, an inhibitor of the fatty acids oxidation promoted for the treatment of diabetes mellitus type 2, should be retired of the market for producing heart hypertrophy [34]. The non expression of PPARα reduces the levels of circulating renin in rats THM/PPARαKO like we mentioned it previously [6]. In subjects with diabetes mellitus type 2 and visceral obesity, hypoadiponectinemia, due to its low production for deficient maturated adipocytes, diminishes positive signals for the expression of PPARα effects in hepatocytes, skeletal muscular cells and maybe in the heart muscle. In patients with visceral obesity there is confirmation about the triglycerides exportation and their deposit in distant organs including the heart [35,36]. Therefore, there are reasons to believe that lipotoxicity in patients with diabetes mellitus may be related to low PPARα activity and hypoadiponectinemia, resulting in a facilitated and predominant PPARγ effect, being easy to understand why do people with diabetes have higher risk to develop heart hypertrophy and congestive heart failure when are exposed to prolonged and therapeutic doses of TZD. PPAR β/δ have been studied less and have potential to participate in adapatative responses, growth, reparation and inflammation, and they surely act integrated with PPARα and PPARγ. Recent experiences in diabetic rats have allowed knowing that hyperglycemia activates the intracellular RAS in their hearts, increasing the oxidative stress and developing cardiac fibrosis [37]; being demonstrated increased renin, angiotensinogen and AII in cardiomiocytes of the examined hearts. The RAS activation in these cases could be due among other, to stimulation of transcriptional factors in response to high glucose levels causing oxidative stress, activating protein kinase C (PKC) trough the hexosamines pathway. The question emerges if the renin expressed in the cardiomiocytes were native or imported, through prorenin internalization by a receptor depending of mannose 6 phosphate [38] or by a specific prorenin/renin receptor [39,40], or by internalization of the AII-AT1receptor [41]. Very well known is the fact that fatty acids and glucose are fundamental energetic substrates in the heart and that cardiac metabolism requires the transcriptional control of genes implied in the transport and metabolism of these substrates, reason why the PPAR activity becomes necessary in front to energy situations as those related with hyperglycemia and atherogenic dislipidemia. Therefore the PPAR activity is practically forced in situations where there is a cellular metabolic challenge and knowing that in the obesity and diabetes mellitus type 2, this challenge is mediated by elevation in the offer of FFA and hyperglycemia (being the FFA natural PPAR agonists), the transcriptional activity promoting the renin gene through PPARα o PPARγ may be another possibility. Of being this way, PPAR have an important role in the occurrence of structural modifications mediated by the RAS in the heart and this a admissible explanation to understand that the sustained and enhanced PPARγ agonism with TZD, in patients with diabetes mellitus, has potential to generate changes conducing to congestive heart failure.
2.4. PPAR, RAS and Vascular Wall
It is known until this moment that the amount of renin locally produced by organs and adipose tissue is very little compared to that originated by the juxtaglomerular apparatus of the afferent arterioles [41] that puts renin in direct contact with the vascular torrent. Even if a specific receptor for prorenin/renin has been found in several tissues [39] , there is another receptor that may be glycolsilated and has residues of mannose 6 phosphate that binds to the insulin growth factor receptor 2 (IGF 2R) and has been identified in human organs like the heart and vascular endothelium [42,43]. The binding of prorenin/renin to one or another receptor may result in different functions and it has been suggested that the activation of the IGF 2 receptor (IGF-2R) confers a purifying signals mechanism [44]. There is not available information about the result of a higher glycosilation of protein, like happens in diabetes mellitus, on the binding of prorrenin/renin to the IGF 2R, but there is evidence that the high plasmatic levels of prorrenin are related with vascular complications of the diabetes [45-47]. Additionally it has been demonstrated that the blockade of the union of the prorrenin/renina to its specific receptor has prevented experimental diabetic glomeruloesclerosis independent of the generation of AII [48]. The binding of the renin to the prorenin/renin receptors actives the mitogen-activated protein kinase (MAPK), regulated by extracellular signals (ERK 1 and 2). These pathways of cellular signaling may increase the tissue fibrosis and the cellular hypertrophy. The activation of the cellular signaling seems to be independent of the AII generation [49]. Prorenin had been also able to increase PAI-1 from vascular soft muscular cells [50,51]. In the vessel wall exists a local tissue RAS [52]. Angiotensinogen is expressed in the vessel smooth muscle, allowing that not alone the plasmatic load coming from the liver would be the substrate to convert it to angiontensin I. This conversion facilitated by renin, will finally result in AII. The RAS interacts with the sympathetic nervous system and with the endothelium, through the nitric oxide (NO) production. Insulin has important cardiovascular actions and it participates in the regulation of the vascular tone, constituting a mechanism that couples the metabolic homeostasis with the hemodynamic one. The insulin resistance involved in the diabetes mellitus type 2, starts with the central obesity, bears atherogenic dyslipidemia and generates endothelial dysfunction contributing to the arterial hypertension and atherogenesis. Inside the physiophatological mechanisms that relate all these actions, the PPAR activity becomes a modulator mechanism of cardiovascular effects, well through the insulin, the lipid metabolism or directly, on the RAS activity. Once insulin binds to its membrane receptor (IR) in the skeletal muscle, liver and adipose tissue and tyrosine phosphorylation take place in the insulin receptor substrate (IRS), a series of signals cascade display in the phosphatidylinositol 3-kinasa-dependent insulin-signaling branch pathway (PI3-K/PDK-1/Akt pathway) culminating with endothelial nitric oxide synthase (eNOS) activity and endothelial vasodilatation mediated by NO. When tyrosine phosphorilation doesn’t happen, signals take place for an alternating mitogenic activated protein kinase (MAPK) dependent branch pathway that induces endothelin-1 (ET1) expression, causing vasoconstriction and sending signals for growth and cellular differentiation that involve the pancreas [53].
In human endothelial cells, physiological concentration of insulin activate receptors of insulin (IR) selectively, generating signals that culminate with endothelial mediated vasodilatation, but supraphysiological concentrations also activate insulin growth factor 1 receptor (IGF-1R) which also couples insulin, increasing the NO production. Insulin acutely increases prostaglandin I 1 (PGI 1) in the vascular endothelium and vascular cellular adhesion molecules 1 (VCAM 1), selectin E, and vascular endothelial growth factor (VEGF) in endothelial cells [54]. The activity of the insulin growth factor 1 (IGF-1) is mitogenic and antiapoptotic on the vascular soft muscular cells and stimulates their migration. The potential reductions in IGF-1 effects could result in benefits in pathological conditions such as hypertension and early stages of atherosclerosis formation plaques characterized by hypertrophy/hyperplasy of the soft muscular cells but harmful in other conditions in which loss of these cells may contribute to destabilize atherosclerotic plaques. IGF-1 binds to proteins and in equimolar mode to one of them, IGBP-3, which stimulates the binding of IGF-1 to IGF-1R, but other proteins such as IGB-2 e IGB-5 are inhibitory and reduce the binding of IGF-1 to its receptor and limits its effect [55]. IGF-2 exerts it effect binding to IGF-1R but also to IGF-2R, called mannose 6 phosphate receptor which acts as a kidnapper or clearing of signals to avoid responses mediated by IGF-2 that if were overexpressed may result in hypoglycemia. If the prorenin/ renin to the mannose 6 phosphate receptor binding is capable to induce decreasing of responses mediated by IGF-2 on the IGF-1R, this could reduce signals to deliver ON. In other words, the binding of renin to prorenin/the mannose 6 phosphate receptor could manipulate signals inhibiting IGF-1 activity and its capacity to induce deliverance of NO. AII in its turn will modify the insulin signaling on its receptor way protein kinase Ac (PKAc) and protein tirosin phosphatase 1B (PTP 1B), decreasing signals in the PI3-K/PDK-1/Akt branch pathway to reduce eNOS acivity and avoid the vasodilator effect mediated by ON. The interference caused by AII to reduce the insulin vasodilator activity transcends until pancreatic effects reducing insulin secretion [56], limiting the appropriate maturation of adipocytes from preadipocytes in the visceral adipose tissue with consequent decreasing of the adiponectin production and so impairing its reinforcement on the insulin action. AII produces upward regulation of IGF-1R mRNA [57,58], which puts more than manifest the interaction of the RAS with signaling cascades initiated by insulin, IFG-1 and IGF-2 to influence the vascular tone, cellular growth, apoptosis regulation, metabolic processes and energy homeostasis. The PPAR activity operates these metabolic and hemodynamic processes linked to the RAS and insulin and associates, contributing to express substances that exert regulator functions from RAS local systems such as visceral adipocytes. This way, PPARγ are regulating operators of the insulin secretion and its tissue, vascular and metabolic actions (vasodilator effect, liver neoglucogenesis, glucose homeostasis, thermogenesis, lipogenesis, lipolysis, regulation of the alimentary intake and adipocyte differentiation among others), working to reinforce insulin signaling trough PI3-K branch pathway and opposing to insulin effects trough MAPK branch pathway as well as AII effects mediated by receptors AT1 and maybe forwards by Angiotensin IV (A IV) on insulin regulated aminopeptidase-Angiotensin IV receptor (IRAP AT4 receptor). The PPARγ activity also opposes to the atherothrombotic effects of the AII in the arterial wall by several control mechanisms counteracting the increment of the plasminogen activator inhibitor-1 (PAI-1), on the VEFG, COX2 expression, platelet activation, oxidative stress, inflammatory effects mediated by TNFα and cytokines, vascular hypertrophy, remodeling, apoptosis and vasoconstriction. To this a decrease of the small and dense LDL cholesterol particles is added, as well a reduction of the venous portal levels of FFA and triglycerides. To favor the PPARγ antiatherogenic action in the arterial wall exists the evidence that the TZD inhibits the formation of the early atherosclerotic lesions in diabetic and nondiabetic mice LDL receptor deficient [59]. PPARα and PPARγ activity may induce signals to express renin in the competent tissues where it is possible, enhancing the local and circulating production of AII. PPARγ activated opposes to the AII effects but may induce renin producing, establishing an mechanism to get a balance as it have been explained before in this review when we talked about visceral adipocytes.
3. Pharmacological Interactions between PPAR and the RAAS and Clinical Implication
It is well known that AT1 receptor blockers are able to activate the PPARγ with differences in their agonist power related with their plasma level, lipophilicity and molecular structure. It has been speculated that telmisartan is one with higher agonist power [60,61]. There are numerous observational studies in which it has been suggested that the AT1 receptor blockers as partial PPARγ agonists have beneficial effects to prevent diabetes mellitus type 2; the NAVIGATOR study was specifically designed to demonstrate this benefit with valsartan [62]. In a same way there are numerous studies that show the benefit of these agents in the reduction or impact of diabetes mellitus complications, such as MARVAL [63], IRMA 2 [64], IDNT [65], RENAAL [66], DETAIL [67], BENEDICT [68], AMADEO [69] and CHARM [70], among others. PPARα and PPARγ (TZD) have also demonstrated benefit in reducing the impact of diabetes on the renal function [71,72]. PPARγ have demonstrated benefit in the reduction of cardiovascular risk factors although in a non overwhelming way in cardiovascular outcomes related with morbidity and mortality. The favored risk factors were: hyperglycemia [73,74], arterial hypertension [75], HDLc [76], LDLc [77], C reactive protein [78], PAI-1 [79], albuminuria/creatinuria index [80] and endothelial function [81]. In the PROACTIVE trial, significant reduction of the relative risk was demonstrated with pioglitazone, in a secondary outcome including mortality of all the causes, non fatal myocardial infarction and stroke, in patients with diabetes mellitus type 2 who had high risk of macrovascular events [82]. There are also studies where the antiatherogenic benefit of TZD was shown in some surrogated outcomes [83]. Recent information allows us to know that in rat hearts with induced diabetes by streptozotoxin, hyperglycemia induces myocardial fibrosis mediated by the RAAS activation which was notably attenuated by aliskiren, a direct renin inhibitor [37]. In cases like this, the expression of renin in cardiomiocytes could be influenced by PPAR α or γ stimulation in response to hyperglycemia or free fatty acids elevation.
4. Perspectives and Conclusions
The RAAS not only intervenes in hidrosalin control and regulation of the vascular tone if not in the modulation of the energy homeostasis and the glucose and lipid metabolism with who maintains very significant iteractions. PPARs act as operators of many mechanisms of the RAAS, especially their local systems linked with the operation of specific tissues and organs [84]. PPAR agonism always bring responses with systemic and local results but PPARα, γ, β/δ are integrated and their individual manipulation is not selective, being able to decompensate the integrated modulation resulting not favorable responses. The experience with dual PPAR agonists as it happened with muraglitazar [85] on cardiovascular mortality in diabetic patients, it is a confirmatory alert about what it can happen when we attempt dual or multiple manipulation committing the modulator interrelations between nuclear receptors, still without considering the potential of tumors and carcinogenesis. The possibility of cardiovascular side effects that has generated so much controversy with PPARγ agonists [86] indicates that we should still learn and investigate more about how to achieve appropriate benefits with them in diabetes and dyslipidemia. Appropriate doses of agonists to use and the profile of the selected patients with diabetes who are indicated would be a key point to choose the best therapy and the best agonist. Meanwhile there are many evidences that a limited PPARγ agonist activity as the AT1 receptor blockers have, especially in diabetes risk or with the established disease, generates important cardiovascular and metabolic beneficial effects. The understanding of that the RAAS couples metabolic and energetic control mechanisms, opens investigation opportunities about the possibility to intervene therapeutically intracellular systems of RAS from renin expression to its binding to receptors due to that potential damage of the target organs starts with the renin generation an before that the AII effects would appear. An excess in blocking the RAAS activity also could affect its control processes such as oxidative stress, mitosis and apoptosis, collagen deposition, inflammatory activity and renal hemodynamics resulting in undesirable outcomes as it has happened in patients with diabetes mellitus type 2 and kidney disease in some clinical trials [87,88].
REFERENCES
- S. Engeli, P. Chling, K. Gorzelniak, et al., “The AdiposeTissue Renin-Angiotensin-Aldosterone System: Role in the Metabolic Syndrome?” The International Journal of Biochemistry & Cell Biology, Vol. 35, No. 6, 2003, pp. 807-825. doi:10.1016/S1357-2725(02)00311-4
- F. Massiera, M. Bloch-Faure, D. Ceiler, et al., “Adipose Angiotensinogen Is Involved in Adipose Tissue Growth and Blood Pressure Regulation,” The FASEB Journal, Vol. 15, No. 14, 2001, pp. 2727-2729.
- L. Yvan-Charvet and A. Quignard-Boulange, “Role of Adipose Tissue Rennin-Angiotensin System in Metabolic and Inflammatory Diseases Associated with Obesity,” Kidney International, Vol. 79, No. 2, 2011, pp. 162-168. doi:10.1038/ki.2010.391
- T. C. Leone, et al., “A Critical Role for the Peroxisome Proliferator Activated Receptor α (PPARα) in the Cellular Fasting Response: The PPARa-Null as a Model of Fatty Oxidation Disorders,” Proceedings of the National Academy of Sciences of USA, Vol. 96, No. 13, 1999, pp. 7473-7478. doi:10.1073/pnas.96.13.7473
- S. Kersten, et al., “Peroxisome Proliferator Activated Receptor α Mediates the Adaptive Response to Fasting,” Journal of Clinical Investigation, Vol. 103, No. 11, 1999, pp. 1489-1498. doi:10.1172/JCI6223
- K. Tordjman, et al., “Absence of Peroxisome ProliferatorActivated Receptor-α Abolishes Hypertension and Attenuates Atherosclerosis in the Tsukuba Hypertensive Mouse,” Hypertension, Vol. 50, No. 5, 2007, pp. 945-951. doi:10.1161/HYPERTENSIONAHA.107.094268
- Ch. Yagil and Y. Yagil, “Peroxisome Proliferator-Activate Receptor-α Friend or Foe?” Hypertension, Vol. 50, No. 5, 2007, pp. 847-850. doi:10.1161/HYPERTENSIONAHA.107.100461
- Y. Shimamoto, K. Hirota and A. Fukamizu, “Effect of Peroxisome Proliferator-Activated Receptor Alpha on Human Angiotensinogen Promoter,” International Journal of Molecular Medicine, Vol. 13, No. 5, 2004, pp. 729- 733.
- H. Yu and R. Di Nicolantonio, “Altered Nuclear Protein Binding to the First Intron of the Renin Gene of the Spontaneously Hypertensive Rat,” Clinical and Experimental Hypertension, Vol. 20, No. 8, 1998, pp. 817-832. doi:10.3109/10641969809053249
- C. Karlsson, K. Lindell, et al., “Human Adipose Tissue Expresses Angiotensinogen and Enzymes Required for Its Conversion to Angiotensina II,” Journal of Endocrinology and Metabolism, Vol. 83, No. 11, 1998, pp. 3925- 3929. doi:10.1210/jc.83.11.3925
- A. Harte, et al., “Insulin-Mediated Upregulation of the Renin Angiotensin System in Human Subcutaneous Adipocytes Is Reduced by Rosiglitazone,” Circulation, Vol. 111, No. 15, 2005, pp. 1954-1961. doi:10.1161/01.CIR.0000161954.17870.5D
- F. J. Morales Olivas and L. Estañ Yago, “Conceptos Nuevos Sobre el Sistema Renina Angiotensina,” Hipertensión y Riesgo Vascular, Vol. 27, No. 5, 2010, pp. 211- 217.
- V. T. Todorov, M. Desch, et al., “Peroxisome Proliferator-Activated Receptor γ Is Involved in the Control of Renin Gene Expression,” Hypertension, Vol. 50, No. 5, 2007, pp. 939-944. doi:10.1161/HYPERTENSIONAHA.107.092817
- V. Todorov, M. Muller, F. Schweda and A. Kurtz, “Tumor Necrosis Factor-Alpha Inhibits Renin Gene Expression,” American Journal of Physiology—Regulatory Integrative and Comparative Physiology, Vol. 283, No. 5, 2002, pp. R1046-R1051.
- L. A. Velloso, et al., “Cross-Talk between the Insulin and Angiotensin Signaling Systems,” Proceedings of the National Academy of Sciences of USA, Vol. 93, No. 22, 1996, pp. 12490-12495. doi:10.1073/pnas.93.22.12490
- M. Furuhashi, N. Ura, K. Higashiura et al., “Blockade of the Renin-Angiotensin System Increases Adiponectin Concentrations in Patients with Essential Hypertension,” Hypertension, Vol. 42, No. 1, 2003, pp. 76-81. doi:10.1161/01.HYP.0000078490.59735.6E
- R. Clasen, M. Schupp, A. Foryst-Ludwig, et al., “PPARgActivating Angiotensin Type-1 Receptor blockers Induce Adiponectin,” Hypertension, Vol. 46, No. 1, 2005, pp. 137-143. doi:10.1161/01.HYP.0000168046.19884.6a
- L. Qlao and J. Shao, “SIRT 1 Regulates Adiponectin Gene Expression through Foxo-C/Enhancer Binding Protein α Transcriptional Complex,” The Journal of Biological Chemistry, Vol. 281, No. 52, 2006, pp. 30015-30034.
- D. Accili, “Lilly Lecture 2003,” Diabetes, Vol. 53, No. 7, 2004, pp. 1633-1642. doi:10.2337/diabetes.53.7.1633
- A. Barthel, “FoxO Proteins in Insulin Action and Metabolism,” Trends in Endocrinology & Met, Vol. 16, No. 4, 2005, pp. 183-189. doi:10.1016/j.tem.2005.03.010
- J. E. Hall, et al., “Obesity Hypertension: Role of Leptin and Sympathetic Nervous System,” American Journal of Hematology, Vol. 14, No. 6, 2001, pp. 103S-115S.
- Y. F. Guan, Ch. Hao, et al., “Thiazolidinediones Expand Body Fluid Volume through PPARγ Stimulation of ENaC-Mediated Renal Salt Absorption,” Nature Medicine, Vol. 11, No. 8, 2005, pp. 861-866. doi:10.1038/nm1278
- K. Tachibana, N. Anzai, et al., “Analysis of PPAR Alpha Function in Human Kidney Cell Line Using siRNA,” Nucleic Acids Symposium Series (Oxford), Vol. 50, No. 1, 2006, pp. 257-258. doi:10.1093/nass/nrl128
- A. C. Calkin, S. Giunti, et al., “PPAR α-γ Agonists Attenuate Diabetic Kidney Disease in the Apolipoprotein E Knockout Mouse,” Nephrology Dialysis Transplantation, Vol. 21, No. 9, 2006, pp. 2399-2405. doi:10.1093/ndt/gfl212
- W. H. Tang, “Do Thiazolidinediones Cause Heart Failure? A Critical Review,” Cleveland Clinic Journal of Medicine, Vol. 73, No. 4, 2006, pp. 390-396. doi:10.3949/ccjm.73.4.390
- S. Farmer, “Transcriptional Control of Adipocyte Formation,” Cell Metabolism, Vol. 4, No. 4, 2006, pp. 263-273. doi:10.1016/j.cmet.2006.07.001
- T. Harsløf, L. Wamberg, et al., “Rosiglitazone Decreases Bone Mass and Bone Marrow Fat,” The Journal of Clinical Endocrinology & Metabolism, Vol. 96, No. 5, 2011, pp. 1541-1548. doi:10.1210/jc.2010-2077
- J. El-Hage, “Peroxisome Proliferator-Activated Receptor (PPAR) Agonists Preclinical and Clinical Cardiac Safety Considerations.” www.fda.gov/downloads/AboutFDA/.../CDER/ucm119071.pdf
- M. Asakawa, H. Takano, T. Nagai, H. Uozumi, et al., “Peroxisome Proliferator-Activated Receptor Gamma Plays a Critical Role in Inhibition of Cardiac Hypertrophy in Vitro and in Vivo,” Circulation, Vol. 105, No. 10, 2002, pp. 1240-1246. doi:10.1161/hc1002.105225
- K. Yamamoto, R. Ohki, R. T. Lee, U. Ikeda, et al., “Peroxisome Proliferator-Activated Receptor Gamma Activators Inhibit Cardiac Hypertrophy in Cardiac Myocytes,” Circulation, Vol. 104, No. 14, 2001, pp. 1670-1675. doi:10.1161/hc4001.097186
- L. Pan and K. W. Gross, “Transcriptional Regulation of Renin: An Update,” Hypertension, Vol. 45, No. 1, 2005, pp. 3-8.
- F. Djouadi, C. J. Weinheimer, J. E. Saffitz, et al., “A Gender-Related Defect in Lipid Metabolism and Glucose Homeostasis in Peroxisome Proliferator Activated Receptor Alpha-Deficient Mice,” Journal of Clinical Investigation, Vol. 102, No. 6, 1998, pp. 1083-1091. doi:10.1172/JCI3949
- Y. Jamshidi, H. E. Montgomery, H. W. Hense, et al., “Peroxisome Proliferator Activated Receptor Alpha Gene Regulates Left Ventricular Growth in Response to Exercise and Hypertension,” Circulation, Vol. 105, No. 8, 2002, pp. 950-955. doi:10.1161/hc0802.104535
- A. Planivila, M. Jové, A. Cabrero and M. Vásquez-Carrera, “Modula el Metabolismo Lipídico del Miocardio la Hipertrofia Cardíaca?” Nefrología, Vol. 24, No. 1, 2004, pp. 29-33.
- J. McGavock, R. G. Victor, R. H. Unger, et al., “Adiposity of the Heart,” Annals of Internal Medicine, Vol. 144, No. 7, 2006, pp. 517-524. doi:10.7326/0003-4819-144-7-200604040-00011
- P. J. Campbell, M. G. Carlson and N. Nurjhan, “Fat Metabolism in Human Obesity,” American Journal of Physiology, Vol. 266, No. 4, 1994, pp. E600-E605.
- V. P. Singh, L. Bao, R. Khode et al., “Intracellular Angiotensin II Production in Diabetic Rats Is Correlated With Cardiomyocyte Apoptosis, Oxidative Stress, and Cardiac Fibrosis,” Diabetes, Vol. 57, No. 12, 2008, pp. 3297-3306. doi:10.2337/db08-0805
- C. A. M. van Kesteren, et al., “Mannosa-6 Phosphate Receptor Mediated Internalization and Activation of Prorenin by Cardiac Cells,” Hypertension, Vol. 30, No. 6, 1999, pp. 1389-1396. doi:10.1161/01.HYP.30.6.1389
- G. Nguyen, F. Delarue, C. Burckle, et al., “Pivotal Role of the Renin/Prorenin Receptor in Angiotensin II Production and Celular Responses to Angiotensin,” Journal of Clinical Investigation, Vol. 109, No. 11, 2002, pp. 1417- 1427.
- T. L. Reudelhuber, “Prorenin, Renin, and Their Receptor: Moving Targets,” Hypertension, Vol. 55, No. 5, 2010, pp. 1071-1074. doi:10.1161/HYPERTENSIONAHA.108.120279
- C. Petrel and E. Clauser, “Angiotensin II AT1 Receptor Constitutive Activation: From Molecular Mechanisms to Pathophysiology,” Molecular and Cellular Endocrinology, Vol. 302, No. 2, 2009, pp. 176-184. doi:10.1016/j.mce.2008.10.049
- R. N. Re, “The Clinical Implication of Tissue Renin Angiotensin Systems,” Current Opinion in Cardiology, Vol. 16, No. 6, 2001, pp. 317-327. doi:10.1097/00001573-200111000-00002
- P. J. Admiraal, et al., “Uptake and Proteolytic Activation of Prorenin by Cultured Human Endothelial Cells,” Journal of Hypertension, Vol. 17, No. 5, 1999, pp. 621-629. doi:10.1097/00004872-199917050-00005
- J. J. Saris, M. M. van den Eijnden, J. M. Lamers, et al., “Prorenin-Induced Myocyte Proliferation: No Role for Intracellular Angiotensin,” Hypertension, Vol. 39, No. 2, 2002, pp. 573-577. doi:10.1161/hy0202.103002
- H. Yokota, T. Nagacoa, et al., “Higher Levels of Prorenin Predict Development of Diabetic Retinopathy in Patients with Type 2 Diabetes,” Journal of the Renin-AngiotensinAldosterone System, Vol. 12, No. 3, 2011, pp. 290-294.
- L. Davies, G. R. Fulcher, A. Atkins, et al., “The Relationship of Prorenin Values to Microvascular Complications in Patients with Insulin Dependent Diabetes Mellitus,” Journal of Diabetes and Its Complications, Vol. 13, No. 1, 1999, pp. 45-51. doi:10.1016/S1056-8727(98)00020-8
- A. A. Franken, F. H. Derkx, A. J. Man in’t Veld, et al., “High Plasma Prorenin in Diabetes Mellitus and Its Correlation with Some Complications,” The Journal of Clinical Endocrinology & Metabolism, Vol. 71, No. 4, 1990, pp. 1008-1015. doi:10.1210/jcem-71-4-1008
- A. Ichihara, F. Suzuki, T. Nakagawa, et al., “Prorenin Receptor Blockade Inhibits Development of Glomerulosclerosis in Diabetic Angiotensin II Type 1a ReceptorDeficient Mice,” Journal of the American Society of Nephrology, Vol. 17, No. 7, 2006, pp. 1950-1961. doi:10.1681/ASN.2006010029
- A. Ichihara, Y. Kaneshiro, T. Takemitsu, et al. “Nonproteolytic Activation of Prorenin Contributes to Development of Cardiac Fibrosis in Genetic Hypertension,” Hypertension, Vol. 47, No. 5, 2006, pp. 894-900. doi:10.1161/01.HYP.0000215838.48170.0b
- H. J. Zhang, N. Noble, W. A. Border, et al., “ReceptorDependent Prorenin Activation and Induction of PAI-1 Expression in Vascular Smooth Muscle Cells,” American Journal of Physiology—Endocrinology and Metabolism, Vol. 295, No. 4, 2008, pp. E810-E819. doi:10.1152/ajpendo.90264.2008
- S. M. Nicholl, E. Roztocil and M. G. Davies, “Plasminogen Activator System and Vascular Disease,” Current Vascular Pharmacology, Vol. 4, No. 2, 2006, pp. 101-116. doi:10.2174/157016106776359880
- V. J. Dzau, “Theodore Cooper Lecture: Tissue Angiotensin and Pathobiology of Vascular Disease: A Unifying Hypothesis,” Hypertension, Vol. 37, No. 4, 2001, pp. 1047-1052. doi:10.1161/01.HYP.37.4.1047
- R. Muniyappa, M. Montagnani, K. Kon Koh, et al., “Cardiovascular Actions of Insulin,” Endocrine Reviews, Vol. 28, No. 5, 2007, pp. 463-491. doi:10.1210/er.2007-0006
- M. Motagnani, I. Golychenco, et al., “Inhibition of Phosphatidylinositol 3-Kinasa Enhances Mitogenic Actions of Insulin in Endothelial Cells,” The Journal of Biological Chemistry, Vol. 277, No. 3, 2002, pp. 1794-1799. doi:10.1074/jbc.M103728200
- P. Delafontaine, Y. H. Song and Y. Li, “Expression, Regulation, and Function of IGF-1, IGF-1R, and IGF-1 Binding Proteins in Blood Vessels,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 24, No. 3, 2005, pp. 435-444. doi:10.1161/01.ATV.0000105902.89459.09
- D. Fliser, F. Schaefer, et al., “Angiotensin II Affects Basal, Pulsatile and Glucose-Stimulated Insulin Secretion in Humans,” Hypertension, Vol. 30, No. 5, 1997, pp. 1156-1161. doi:10.1161/01.HYP.30.5.1156
- K. J. Scheidegger, J. Du and P. Delafontaine, “Distinct and Common Pathways in the Regulation of Insulin-Like Growth Factor-1 Receptor Gene Expression by Angiotensin II and Basic Fibroblast Growth Factor,” The Journal of Biological Chemistry, Vol. 274, No. 6, 1999, pp. 3522-3530. doi:10.1074/jbc.274.6.3522
- P. Delafontaine and H. Lou, “Angiotensin II Regulates Insulin-Like Growth Factor I Gene Expression in Vascular Smooth Muscle Cells,” The Journal of Biological Chemistry, Vol. 268, No. 22, 1993, pp. 16866-16870.
- A. R. Collins, W. P. Meehan, U. Kintscher, et al., “Troglitazone Inhibits Formation of Early Atherosclerotic Lesions in Diabetic and Nondiabetic Low Densitity Lipoprotein Receptor-Deficient Mice,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 21, No. 3, 2001, pp. 365-371. doi:10.1161/01.ATV.21.3.365
- M. Schupp, J. Janke, R. Claren, T. Unger, et al., “Angiotensin Type 1 Receptor Blockers Induce Peroxisome Proliferator-Activated Receptor-Gamma Activity,” Circulation, 2004; Vol. 109, No. 17, pp. 2054-2057. doi:10.1161/01.CIR.0000127955.36250.65
- S. C. Benson, H. A. Pershadsingh, A. Chittiboyina, P. Desai, M. Pravenec, N. Qi, J. Wang, M. A. Avery and T. W. Kurtz, et al., “Identification of Telmisartan as a Unique Angiotensin II Receptor Antagonist with Selective PPAR-Gamma-Modulating Activity,” Hypertension, Vol. 43, No. 5, 2004, pp. 993-1002. doi:10.1161/01.HYP.0000123072.34629.57
- J. McMurray, et al., “Effect of Valsartan on the Incidence of Diabetes and Cardiovascular Events,” The New England Journal of Medicine, Vol. 362, No. 16, 2010, pp. 1477-1490. doi:10.1056/NEJMoa1001121
- G. Viberti and N. M. Wheeldon, “Microalbuminuria Reduction with Valsartan in Patients with Type 2 Diabetes Mellitus,” Circulation, Vol. 106, No. 6, 2002, pp. 672- 678. doi:10.1161/01.CIR.0000024416.33113.0A
- H. H. Parving, H. Lehnert, J. Brochner-Mortensen, et al., “The Effect or Irbesartan on the Development of Diabetic Nephropathy in Patients with Type 2 Diabetes,” The New England Journal of Medicine, Vol. 345, No. 12, 2001; pp. 870-878. doi:10.1056/NEJMoa011489
- E. J. Lewis, L. G. Hunsicker, et al., “Collaborative Study Group, Irbesartan diabetic nephropathy trial (IDNT),” The New England Journal of Medicine, Vol. 345, 2001, pp. 851-860. doi:10.1056/NEJMoa011303
- B. Brenner, M. Cooper, D. Zeeuw, et al., “Effects of Losartan on Renal and Cardiovascular Outcomes in Type 2 Diabetes and Nephropathy,” The New England Journal of Medicine, Vol. 345, No. 12, 2001, pp. 861-869. doi:10.1056/NEJMoa011161
- A. H. Barnett, S. C. Bain, et al., “Angiotensin-Receptor Blockade versus Converting-Enzyme Inhibition in Type 2 Diabetes and Nephropathy,” The New England Journal of Medicine, Vol. 351, No. 19, 2004, pp. 1952-1961. doi:10.1056/NEJMoa042274
- P. Ruggenenti, A. Fassi, et al., “Preventing Microalbuminuria in Type 2 Diabetes,” The New England Journal of Medicine, Vol. 351, No. 19, 2004, pp. 1941-1951. doi:10.1056/NEJMoa042167
- G. Bakris, E. Burgess, M. Weir, et al., “Telmisartan Is More Effective than Losartan in Reducing Proteinuria in Patients with Diabetic Nephropathy,” Kidney International, Vol. 74, No. 3, 2008, pp. 364-369. doi:10.1038/ki.2008.204
- S. Yusuf, J. Ostergren, H. Gerstein, et al., “Effects of Candesartan on the Development of a New Diagnosis of Diabetes Mellitus in Patients With Heart Failure,” Circulation, Vol. 112, No. 1, 2005, pp. 48-53. doi:10.1161/CIRCULATIONAHA.104.528166
- G. Bakris, et al., “Rosiglitazone Reduces Urinary Albumin Excretion in type 2 Diabetes,” Journal of Human Hypertension, Vol. 17, No. 1, 2003, pp. 7-12. doi:10.1038/sj.jhh.1001444
- A. C. Calkin, S. Giunti, K. A. Jandeleit-Dahm, et al., “PPAR-alpha and -gamma Agonists Attenuate Diabetic Kidney Disease in the Apolipoprotein E Knockout Mouse,” Nephrology Dialysis Transplantation, Vol. 21, No. 9, 2006, pp. 2399-2405. doi:10.1093/ndt/gfl212
- C. Rosak, R. Petzoldt, R. Wolf, et al., “Rosiglitazone Plus Metformin is Effective and Well Tolerated in Clinical Practice. Results from Large Observational Studies in People with Diabetes,” International Journal of Clinical Practice, Vol. 59, No. 10, 2005, pp. 1131-1136. doi:10.1111/j.1368-5031.2005.00652.x
- L. Reynolds, E. Konz, R. Frederich, et al., “Rosiglitazone Amplifies the Benefits of Lifestyle Intervention Measures in Long-Standing Type 2 Diabetes Mellitus,” Diabetes, Obesity and Metabolism, Vol. 4, No. 4, 2002, pp. 270- 275. doi:10.1046/j.1463-1326.2002.00207.x
- M. St. John Sutton, M. Rendell, P. Dandona, et al., “A Comparison of the Effects of Rosiglitazone and Glyburide on Cardiovascular Function and Glycemic Control in Patients with Type 2 Diabetes,” Diabetes Care, Vol. 25, No. 11, 2002, pp. 2058-2064. doi:10.2337/diacare.25.11.2058
- H. E. Lebovitz, J. F. Dole, R. Patwardhan, et al., “Rosiglitazone Montherapy Is Effective in Patients with Type 2 Diabetes,” The Journal of Clinical Endocrinology & Metabolism, Vol. 86, No. 1, 2001, pp. 280-288. doi:10.1210/jc.86.1.280
- M. I. Freed, R. Ratner, S. M. Marcovina, et al., “Effects of Rosiglitazone Alone and in Combination with Atorvastatin on the Metabolic Abnormalities in Type 2 Diabetes Mellitus,” American Journal of Cardiology, Vol. 90, No. 9, 2002, pp. 947-952. doi:10.1016/S0002-9149(02)02659-0
- S. M. Haffner, A. S. Greenberg, W. M. Weston, et al., “Effect of Rosiglitazone Treatment on Nontraditional Markers of Cardiovascular Disease in Patients with Type 2 Diabetes Mellitus,” Circulation, Vol. 106, No. 6, 2002, pp. 679-684. doi:10.1161/01.CIR.0000025403.20953.23
- N. Marx, T. Bourcier, G. K. Sukhova, et al., “PPARγ Activation in Human Endothelial Cells Increases Plasminogen Activator Inhibitor Type-1 Expression. PPARγ as a Potential Mediator in Vascular Disease,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 19, No. 3, 1999, pp. 546-551. doi:10.1161/01.ATV.19.3.546
- G. Bakris, G. Viberti, W. M. Weston, et al., “Rosiglitazone Reduces Urinary Albumin Excretion in Type 2 Diabetes,” Journal of Human Hypertension, Vol. 17, No. 1, 2003, pp. 7-12.
- A. Natali, S. Baldeweg, E. Toschi, B. Capaldo, D. Barbaro, et al., “Vascular Effects of Improving Metabolic Control with Metformin or Rosiglitazone in Type 2 Diabetes,” Diabetes Care, Vol. 27, No. 6, 2004, pp. 1349- 1357. doi:10.2337/diacare.27.6.1349
- J. Dormandy, B. Charbonnel, D. Eckland, et al., “Secondary Prevention of Macrovascular Events in Patients with Type 2 Diabetes in the PROactive Study (Prospective Pioglitazone Clinical Trial in Macrovascular Events): A Randomized Controlled Trial,” Lancet, Vol. 366, No. 9493, 2005, pp. 1279-1289. doi:10.1016/S0140-6736(05)67528-9
- S. Nissen, S. Nicholls, K. Wolski, R. Nesto, et al., “Comparison of Pioglitazone vs Glimepiride on Progression of Coronary Atherosclerosis in Patients with Type 2 Diabetes: The PERISCOPE Randomized Controlled Trial,” JAMA, Vol. 299, No. 13, 2008, pp. 1561-1573. doi:10.1001/jama.299.13.1561
- I. Kuipers, P. van der Harst, G. Navis, et al., “Nuclear Hormones Receptors as Regulators of the Renina-Angiotensin-Aldosterone System,” Hypertension, Vol. 51, No. 6, 2008, pp. 1442-1448. doi:10.1161/HYPERTENSIONAHA.107.108530
- S. Nissen, K. Wolski and E. Topol, “Effect of Muraglitazar on Death and Major Adverse Cardiovascular Events in Patients with Type 2 Diabetes Mellitus,” JAMA, Vol. 294, No. 20, 2005, pp. 1-6. doi:10.1001/jama.294.20.joc50147
- S Nissen and K. Wolski, “Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes,” The New England Journal of Medicine, Vol. 356, No. 24, 2007, pp. 2457-2471. doi:10.1056/NEJMoa072761
- J. F. Mann, R. E. Schmieder, M. McQueen, et al., “Renal Outcomes with Telmisartan, Ramipril or Both, in People at High Vascular Risk (the ONTARGET Study): A Multicentre, Radomised, Double-Blind Controlled Trial,” Lancet, Vol. 372, No. 9638, 2008, pp. 547-553.
- H. H. Parving, B. Brenner, J. J. McMurray, et al., “Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE),” Nephrology Dialysis Transplantation, Vol. 34, No. 5, 2009, pp. 1663-1671. doi:10.1093/ndt/gfn721