Open Journal of Pathology
Vol.2 No.1(2012), Article ID:16944,5 pages DOI:10.4236/ojpathology.2012.21001
Gain of Chromosomes 1, 2, 7, 10, 13 and 17 in an Acquired Cystic Kidney Disease Associated Renal Cell Carcinoma
![]()
1Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, Philadelphia, USA; 2Division of Anatomic and Molecular Pathology, Department of Pathology and Immunology, Washington University School of Medicine, Saint Louis, USA; 3Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, Philadelphia, USA.
Email: *bingz@uphs.upenn.edu
Received October 25th, 2011; revised December 12th, 2011; accepted December 25th, 2011.
Keywords: ACDK-RCC; Oxalate; Interphase in Situ Hybridization
ABSTRACT
The acquired cystic disease of the kidney-associated renal cell carcinoma (ACDK-RCC) in the current study occurred in a kidney with multiple cysts and was composed of cells with eosinophilic cytoplasm and prominent nucleoli. There were extensive calcium oxalate deposits in both non-neoplastic cysts and tumor. The tumor cells were positive for RCC Ma, CD10, and EMA, focally positive for CK7, negative for vimentin. Interphase in situ hybridizations (FISH) were performed for chromosome 1, 2, 7, 10, 13 and 17. No chromosomal abnormality was observed in the non-neoplastic cysts. Polysomies of chromosomes 1, 2, 7, 10, 13, 17 were observed in the tumor. Trisomy 13 was first reported in this type of tumor, which warranted further study.
1. Introduction
Acquired cystic disease of kidney-associated renal cell carcinoma is a renal cancer not included in 2004 WHO classification of renal tumor. Chromosomal analysis in this type of tumor has been reported only in few cases [1-4]. We reported here an ACDK-RCC with a unique chromosome profile, which was not reported previously.
2. Clinical Information and Methods
A 60 year old female patient had a history of systemic lupus erythematosus and renal failure. She had been on daily peritoneal dialysis for 10 hours per day since 1991. MRI showed a 1.9 × 1.5 cm mass in the upper pole of left kidney. A radical nephrectomy was performed.
The specimen was processed routinely. Immunohistochemical studies were performed by using antibodies against vimentin (Mouse monoclonal, 1:60 dilution, Dako, CA), RCC Ma (Mouse monoclonal, 1:250, Neomarkers/Lab Vision, CA), CD10 (Mouse monoclonal, 1:40, Novacastra/- Leica Microsystems, IL), CK7 (Mouse monoclonal, 1:200, Dako, CA), EMA (Mouse monoclonal, 1:100, Dako, CA) and CD57 (Mouse monoclonal, 1:20, Neomarkers/Lab Vision, CA) on Bond III automatic staining system (Leica Microsystems-Buffalo Grove, IL).
Interphase fluorescence in situ hybridization (FISH) was performed on formalin fixed paraffin-embedded tissue sections cut at a thickness of 5-um according to manufacturer’s instruction. Probes used were centromeric specific probes (CEP) for chromosomes 1, 2, 7, 10, and 17 as well as a locus specific (LSI) probe for RB1 (13q14). All probes were from Abbott Molecular, Inc. (Des Plaines, IL). For this study, CEP1 (SpectrumOrange) was paired with CEP10 (SpectrumGreen), CEP2 (SpectrumOrange) was paired with CEP 17(SpectrumGreen) and RB1 (SpectrumOrange) was paired with CEP7 (SpectrumGreen).
3. Interpretation of Fish Results
A minimum of 200 cells were counted for each tissue type and for each probe type. A target tissue was considered to have a monosomy (chromosomal loss) for a particular chromosome if the percentage of nuclei with only 1 signal for the probe was greater than 31%, 33%, 28%, 29%, 29%, or 33% for chromosomes 1, 2, 7, 10, 13, and 17, respectively. Polysomy (chromosomal gain) was found for a particular chromosome if the percentage of nuclei with 3 or more signals for the probe was greater than 10%, for chromosomes 1, 2, 7, 10, 13, and 17. The percentage of cells needed to consider a chromosomal loss or gain was established from a 95% confidence interval utilizing a reference range of normal cells.
4. Pathology
Grossly, the kidney was multicystic. A cystic mass was identified on the upper pole measuring 2.9 × 2.6 × 1.9 cm. The mass was circumscribed by a thick fibrous capsule.
Microscopically, the tumor showed microcytic, tubular and papillary architectures with extensive intratubular and intercellular calcium oxalate deposition (Figures 1(b) and (d); Figure 1(e) shows the higher magnification of 1(b)). The oxalate crystals were also identified in the non-neoplastic cystic areas of the kidney (Figures 1(a) and (c)). The amount of oxalate crystals was graded 3+ in both tumor and nonneoplastic cystic areas [5]. The tumor cells had eosinophilic cytoplasm and prominent nucleoli (Fuhrman grade III).

Figure 1. Non-neoplastic cyst and ACDK-RCC. (a)-(d) non-neoplastic cysts. (a) Non-neoplastic cyst with calcium oxalate, H & E, × 50; (c) oxalate crystals under polarization in non-neoplastic cyst, × 50; (b) ACDK-RCC, H & E, × 50; (d) oxalate crystals under polarization in ACDK-RCC, × 50; (e)-(h) ACDK-RCC; (e) H & E, × 200; (f), RCC, × 200; (g) CK7, × 200; (h) CD57, × 200.
Immunohistochemistry was performed with adequate controls. The tumor cells were positive for RCC Ma (Figure 1(f)), CD10, EMA and CD57 (Figure 1(h)), focally positive for CK7 (Figure 1(g)) and negative for vimentin.
Interphase FISH was performed in both cystic non-neoplastic parenchyma (Figure 2(a)) and tumor (Figure 3(a)) (see Table 1). Chromosome 1, 2 and 13 are labeled as spectrum orange and chromosome 7, 10 and 17 are labeled as spectrum green. The chromosome pairs are as follows in one panel: chromosome 1 and 10 (Figure 2(b) and Figure 3(b)), chromosome 2 and 17 (Figure 2(c) and Figure 3(c)), and chromosome 7 and 13 (Figure 2(d) and Figure 3(d)). No chromosomal abnormality was seen in the non-neoplastic cysts (Figures 2(b), (c) and (d)). The tumor showed numeric abnormalities of chromosome 1, 2, 7, 10, 13, and 17 (Figures 3(b), (c) and (d)). The tumor cells were positive for both trisomy and terasomy for chromosome 7 and 17 (Figures 3(c) and (d)). In addition, there were also trisomies of chromosome 1, 2, and 10 (Figures 3(a) and (c)). Interestingly, the most common chromosome changes were trisomy 13 (in 54% of tumor cells, Figure 3(d)), which were not described in the previous literature.
5. Discussion
Oxalate crystals are observed in about 30% of ACDKRCCs [5-7]. The role of calcium oxalate in the tumorigenesis of this type of cancer is uncertain. We observed the oxalate deposition in both non-neoplastic cysts and tumor, consistent with previous observation [6]. We also found that both tumor and non-neoplastic cysts were positive for CD57, a marker of the ascending loop of Henle [8]. This observation suggests that oxalate deposition in both tumor and non-neoplastic cysts appear to be a result of expression of the Henle’s loop characteristics [6].
Papillary RCCs frequently occur in the dialyzed kidneys [9]. Atypical epithelial proliferations harbor some features of sporadic papillary RCC and therefore may represent early neoplastic lesions [10]. Even more, gains of chromosome 7 and 17 and loss of chromosome Y has been demonstrated in the hyperplasic and dysplastic tubules in non-neoplastic tissue of end-stage kidneys [11], which may contribute to the increased incidence of papillary RCCs in the patients with long-term hemodialysis. As we observed that calcium oxalate crystals were deposited in both non -neoplastic cysts and tumor, FISH for chromosome 1, 2, 7, 10, 13, and 17 were performed on both non-neoplastic renal cysts and tumor to see whether there were differences in chromosomal changes in these areas. There were no significant numeric chromosomal changes in the non-neoplastic cysts, however, polysomies of chromosomes 1, 2, 7, 10, 13, 17 were observed in the tumor. These observations suggest that oxalate deposition may not be part of etiology for the ACDK-RCC [6].
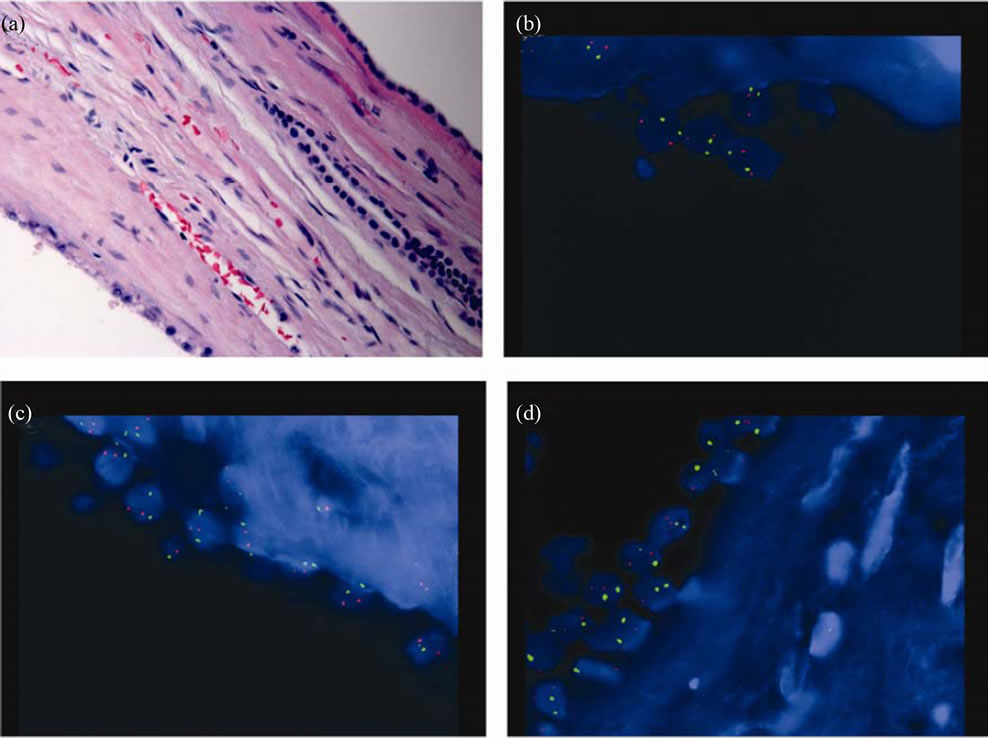
Figure 2. FISH studies for non-neoplastic cyst. (a) H & E, × 400; (b) chromosome 1 (spectrum orange) and 10 (spectrum green), × 1000; (c) chromosome 2 (spectrum orange) and 17 (spectrum green), × 1000; (d) chromosome 13 (spectrum orange) and 7 (spectrum green), × 1000.
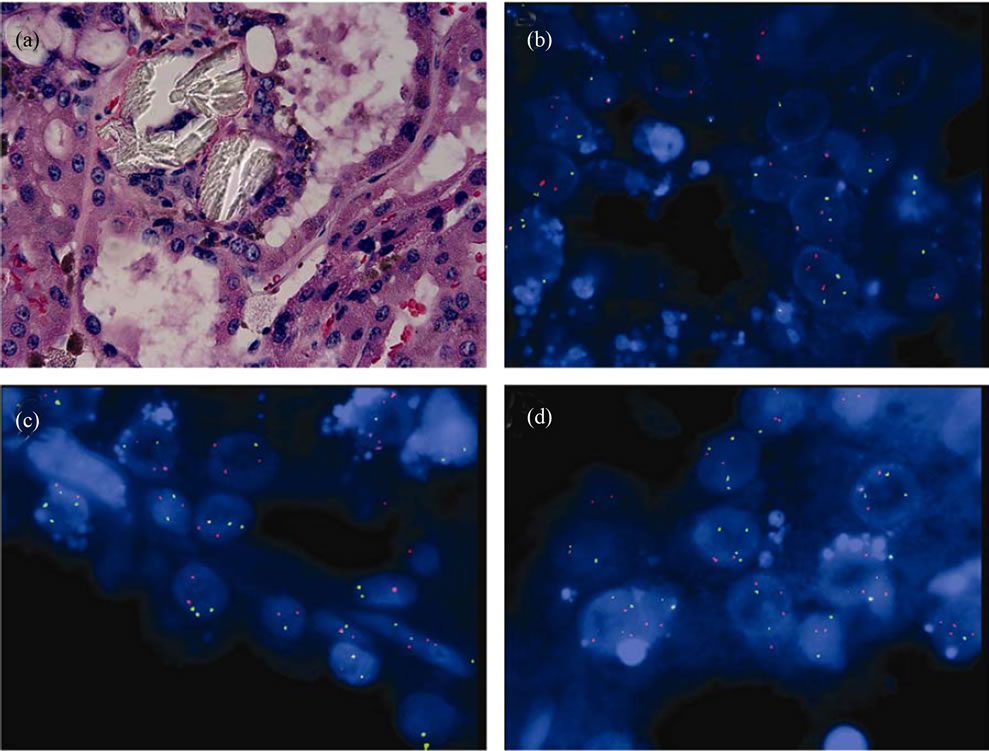
Figure 3. FISH studies for ACDK-RCC. (a) H & E, × 400; (b), chromosome 1 (spectrum orange) and 10 (spectrum green), × 1000; (c) chromosome 2 (spectrum orange) and 17 (spectrum green), × 1000; (d) chromosome 13 (spectrum orange) and 7 (spectrum green), × 1000.
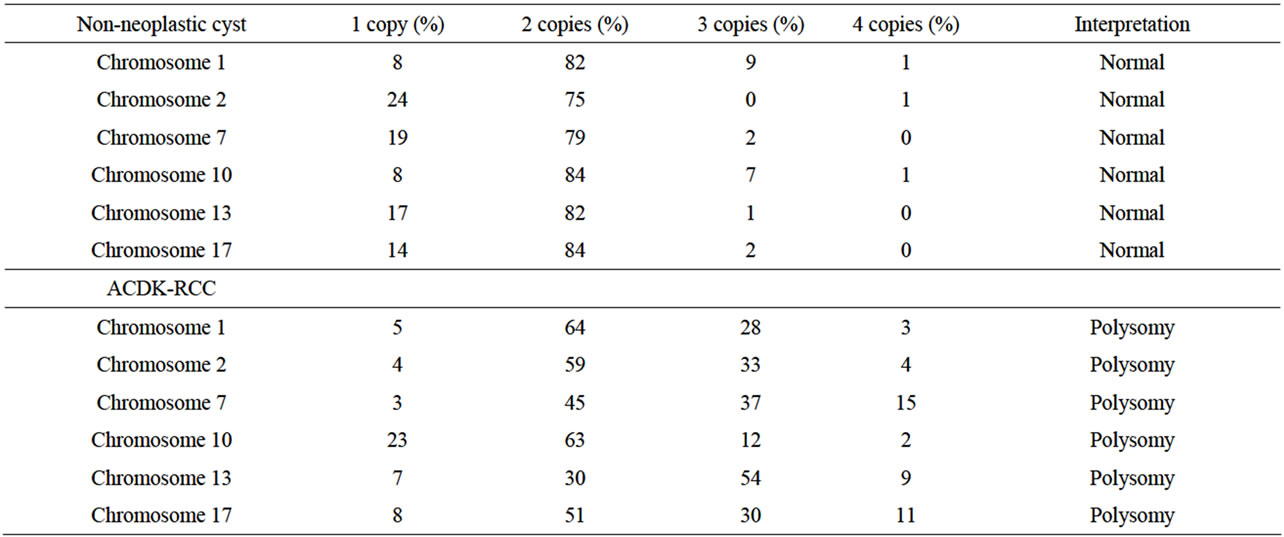
Table 1. FISH studies of non-neoplastic cysts and ACDK-RCC.
Kuroda et al performed G band karyotyping of a ACDKRCC and showed a karyotype of 49, X, +X, –Y, +3, +7, +16 in the tumor, which was similar to those of sporadic papillary RCC [2]. Pan et al performed combined comparative genomic hybridization and FISH analyzed in ACDKRCC and found the common gain of chromosomes 7 and 17 [4]. Our findings of trisomy and tetrasomy of chromosome 7 and 17 are in line with these previous observations and suggest that ACDK-RCC and sporadic papillary RCC may share some common chromosome abnormalities.
Cossu-Recca et al studied changes of chromosome 1, 2, 6, 10, and 17 in three ACDK-RCCs using FISH and found gains of chromosomes 1, 2 and 6 in two tumors and gain of chromosome 10 in one tumor [1]. We also observed polysomies of chromosome 1, 2, 10. Duplication of chromosome 1p was suggested to mark fatal disease progression in papillary RCC [12]. KIF14 and NEK2 might be the target genes [13] and both genes were preferentially expressed in papillary RCCs with fatal outcome [12].
In addition, we also observed the trisomy 13 in this type of tumor, which was not reported previously. Numeric changes in chromosome 13 has been reported in two hereditary papillary RCCs [14] and one familial RCC [15]. The function of gain of chromosome 13 in this case was unclear, further investigation of such change using large cohort is warranted.
Immunohistochemically we observed that the tumor cells were positive for RCC Ma, CD10, focally positive for CK7 and negative for vimentin. This observations were similar to the findings of Kuroda et al [2] and showed similar immunohistochemical profiles between ACDK-RCCs and sporadic papillary RCCs.
In summary we reported a case of ACDK-RCC with a unique chromosome profile and with some similarity to the sporadic RCC.
REFERENCES
- P. Cossu-Rocca, J. N. Eble, S. Zhang, G. Martignoni, M. Brunelli and L. Cheng, “Acquired Cystic Disease-Associated Renal Tumors: An Immunohistochemical and Fluorescence in Situ Hybridization Study,” Modern Pathology, Vol. 19, No. 6, 2006, pp. 780-787.
- N. Kuroda, T. Shiotsu, O. Hes, M. Michal, T. Shuin, G. H. Lee, “Acquired Cystic Disease-Associated Renal Cell Carcinoma with Gain of Chromosomes 3, 7, and 16, Gain of Chromosome X, and Loss of Chromosome Y,” Medical Molecular Morphology, Vol. 43, No. 4, 2010, pp. 231-234. doi:10.1007/s00795-009-0465-8
- N. Kuroda, M. Tamura, N. Hamaguchi, S. Mikami, C. C. Pan, M. Brunelli, G. Martignoni, O. Hes, M. Michal and G. H. Lee, “Acquired Cystic Disease-Associated Renal Cell Carcinoma with Sarcomatoid Change and Rhabdoid Features,” Annals of Diagnostic Pathology, Vol. 15, No. 6, 2011, pp. 462-466. doi:10.1016/j.anndiagpath.2010.07.008
- C. C Pan, Y. J. Chen, L. C. Chang, Y. H. Chang and D. M. Ho, “Immunohistochemical and Molecular Genetic Profiling of Acquired Cystic Disease-Associated Renal Cell Carcinoma,” Histopathology, Vol. 55, No. 2, 2009, pp. 145-153. doi:10.1111/j.1365-2559.2009.03361.x
- N. Sule, U. Yakupoglu, S. S. Shen, B. Krishnan, G. Yang, S. Lerner, D. Sheikh-Hamad and L. D. Truong, “Calcium Oxalate Deposition in Renal Cell Carcinoma Associated with Acquired Cystic Kidney Disease: A Comprehensive Study,” American Journal of Surgical Pathology, Vol. 29, No. 4, 2005, pp. 443-451. doi:10.1097/01.pas.0000152131.58492.97
- Y. Enoki, G. Katoh, H. Okabe and A. Yanagisawa, “Clinicopathological Features and CD57 Expression in Renal Cell Carcinoma in Acquired Cystic Disease of the Kidneys: With Special Emphasis on a Relation to the Duration of Haemodialysis, the Degree of Calcium Oxalate Deposition, Histological Type, and Possible Tumorigenesis,” Histopathology, Vol. 56, No. 3, pp. 384- 394. doi:10.1111/j.1365-2559.2010.03480.x
- S. K. Tickoo, M. N. DePeralta-Venturina, L. R. Harik, H. D. Worcester, M. E. Salama, A. N. Young, H. Moch and M. B. Amin, “Spectrum of Epithelial Neoplasms in End-Stage Renal Disease: An Experience from 66 Tumor-Bearing Kidneys with Emphasis on Histologic Patterns Distinct from Those in Sporadic Adult Renal Neoplasia,” American Journal of Surgical Pathology, Vol. 30, No. 2, 2006, pp. 141-153. doi:10.1097/01.pas.0000185382.80844.b1
- Y. Allory, F. Commo, L. Boccon-Gibod, M. Sibony, P. Callard, P. Ronco and H. Debiec, “Sulfated HNK-1 Epitope in Developing and Mature Kidney: A New Marker for Thin Ascending Loop of Henle and Tubular Injury in Acute Tubular Necrosis,” Journal of Histochemistry & Cytochemistry, Vol. 54, No. 5, 2006, pp. 575-584. doi:10.1369/jhc.5A6791.2006
- I. Ishikawa and G. Kovacs, “High Incidence of Papillary Renal Cell Tumours in Patients on Chronic Haemodialysis,” Histopathology, Vol. 22, No. 2, 1993, pp. 135-139. doi:10.1111/j.1365-2559.1993.tb00091.x
- W. Cheuk, E. S. Lo, A. K. Chan and J. K. Chan, “Atypical Epithelial Proliferations in Acquired Renal Cystic Disease Harbor Cytogenetic Aberrations,” Human Pathology, Vol. 33, No. 7, 2002, pp. 761-765. doi:10.1053/hupa.2002.125370
- O. Hes, R. Sima, J. Nemcova, M. Hora, S. Bulimbasic, D. V. Kazakov, T. Urge, T. Reischig, M. Dvorak and M. Michal, “End-Stage Kidney Disease: Gains of Chromosomes 7 and 17 and Loss of Y Chromosome in NonNeoplastic Tissue,” Virchows Arch, Vol. 453, No. 4, 2008, pp. 313-319. doi:10.1007/s00428-008-0661-2
- A. Szponar, D. Zubakov, J. Pawlak, A. Jauch and G. Kovacs, “Three Genetic Developmental Stages of Papillary Renal Cell Tumors: Duplication of Chromosome 1q Marks Fatal Progression,” International Journal of Cancer, Vol. 124, No. 9, 2009, pp. 2071-2076. doi:10.1002/ijc.24180
- T. W. Corson, A. Huang, M. S. Tsao and B. L. Gallie, “KIF14 is a Candidate Oncogene in the 1q Minimal Region of Genomic Gain in Multiple Cancers,” Oncogene, Vol. 24, No. 30, 2005, pp. 4741-4753. doi:10.1038/sj.onc.1208641
- E. Prat, M. Bernues, J. Del Rey, J. Camps, I. Ponsa, F. Algaba, J. Egozcue, M. R. Caballin, A. Gelabert and R. Miro, “Common Pattern of Unusual Chromosome Abnormalities in Hereditary Papillary Renal Carcinoma,” Cancer Genetics and Cytogenetics, Vol. 164, No. 2, 2006, pp. 142-147. doi:10.1016/j.cancergencyto.2005.09.010
- H. J. Decker, B. Wullich, J. M. Whaley, G. Herrera, S. M. Klauck, A. A. Sandberg, D. W. Yandell and B. R. Seizinger, “Cytogenetic and Molecular Studies of a Familial Renal Cell Carcinoma,” Cancer Genetics and Cytogenetics, Vol. 63, No. 1, 1992, pp. 25-31. doi:10.1016/0165-4608(92)90059-H
NOTES
*Corresponding author.