Open Journal of Biophysics
Vol.3 No.1(2013), Article ID:27486,12 pages DOI:10.4236/ojbiphy.2013.31001
Modeling the Structure of Yeast MATα1: An HMG-Box Motif with a C-Terminal Helical Extension
1Department of Chemistry and Biochemistry, Huntingdon College, Montgomery, USA
2Department of Microbiology, Alabama State University, Montgomery, USA
3School of Public Health, University of Alabama, Birmingham, USA
Email: doba.jackson@huntingdon.edu
Received September 25, 2012; revised October 30, 2012; accepted November 11, 2012
Keywords: MATα1; MATα2; Gene Regulation; Mating-Type; Yeast; α-Domain; Combinatorial Control of Transcription
ABSTRACT
The yeast MATα1 is required for the activation of α-specific genes in Saccharomyces cerevisiae and thus confers the α-cell identity of the yeast. MATα1 contains a domain called the α-domain which has significant sequence identity to the HMG-box family of proteins. A multiple sequence alignment of several α-domains and various structurally determined HMG-box domains have revealed that both domains possess very similar structural and functional residues. We found that the basic amino acids of the N-terminal loop, the intercalating hydrophobic residues of the first helix, and the hydrophobic residues required for interactions within the core of the protein are remarkably conserved in α-domains and HMG-box proteins. Our generated molecular models suggest that the first and third helix will be shorter and that the HMG-box core is not an isolated domain. The region beyond the conserved HMG-box motif contains an extended helical region for about 20 - 30 amino acids. Structural models generated by comparative modeling and ab initio modeling reveal that this region will add two or more additional α-helices and will make significant contacts to helix III, II and I of the HMG-box core. We were able to illustrate how the extended α-domain would bind to DNA by merging of the α-domain and the LEF-1/DNA complex. The models we are reporting will be helpful in understanding how MATα1 binds to DNA with its partner MCM1 and activates transcription of α-specific genes. These models will also aid in future biophysical studies of MATα1 including the crystallization and structure determination.
1. Introduction
The sex-determining genes of fungi reside at one or two specialized regions of the chromosome and are known as the mating-type (MAT) loci. The expression of the genes on the MAT loci is sufficient to confer haploid cell identity, attract compatible mating partners and prepare the cell for sexual reproduction (reviewed in [1-4]). A number of MAT loci have been described in ascomycetes and basidiomycetes [5]. Bioinformatic and structural analysis of the transcription factors have revealed each transcription factor conserved regions that belong to one of three different DNA-binding protein families: 1) the HMG-box superfamily; 2) the Homeodomain superfamily; 3) the α-domain family. In the zygomycetes, the earliest branching fungal lineage characterized so far, each MAT loci of the two cell types both have HMGtype transcription factors. However, in ascomycetes and later branching fungal lineage characterized so far, the MAT loci of the two haploid cell types contain a combination of α-domain, homeodomain and HMG-box transcription factors [1,6]. This leads to the question of what is the evolutionary relationship that exists between these three transcription factor families.
A highly established model of sexual identity is the ascomycete yeast Saccharomyces cerevisiae. S. cereviseae has three mating types (a, α, a/α) which are controlled by a complex array of DNA binding transcription factors (Figure 1) located within the MAT loci (reviewed in [1-4,7]). In the α-cell, the MATα1 transcription factor (an α-domain protein) interacts with a general transcription factor MCM1 protein and upstream activation sequences (UAS) to activate the expression of α-specific genes. MCM1 also interacts with the α2 protein (a homeodomain protein) and the UAS of a-specific genes to repress their transcription in α-cells. MCM1 is a member of the MADS-box family of transcription factors that play pivotal roles in regulating biological processes in a diverse range of eukaryotic organisms [8]. The UAS’s of α-specific and a-specific genes contain sequences that bind the MATα1/MCM1 and the MATα2/
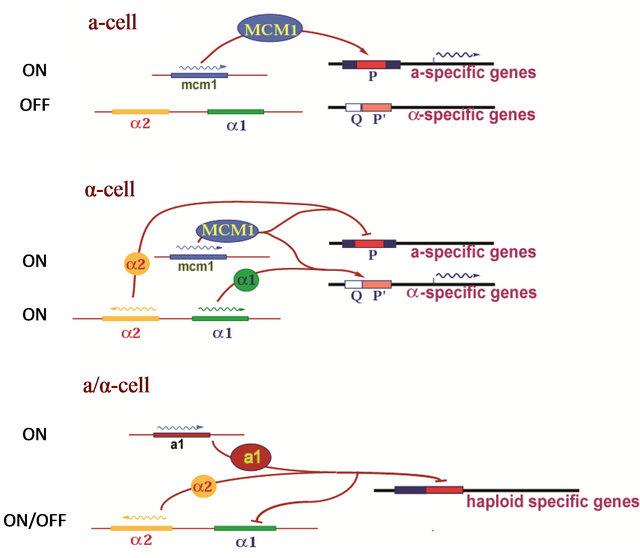
Figure 1. A simple model of mating-type determination in yeast Saccharomyces cerevisiae [1-4].
MCM1 proteins respectively. The crystal structure of the MADS-box region of MCM1 in complex with α2 has been determined to 2.25 Å [9]. In this structure, MCM1 binds DNA as a homodimer and uses a long α-helix to bind and bend the DNA by 72˚. Two α2 homeodomains are spaced on both sides of the MCM1 dimer and make symmetry-related contacts to MCM1 by parallel β-sheets.
The mode of MATα1 binding to DNA and its exact role in transcriptional activation is not known. MATα1 binds MCM1 directly and along a strand of duplex DNA [10]. However, by itself, purified MATα1 is not very efficient in binding to DNA [11]. Many studies have established that the MATα1/MCM1/DNA complex is held together by direct protein-protein interactions and direct interactions with DNA [10-12]. We know that mutations in the Q-element of the QP-box affect the ternary complex formation but, do not affect the weak binding of MCM1 alone. We also know that α1 induced DNA bending is required for transcriptional activation whereas DNA bending by MCM1 alone or by mutations is insufficient [11]. However, despite the abundance of biochemical studies, it is unclear how the MATα1 protein recognizes the QP-box DNA and MCM1 upon activation of transcription of α-specific genes. The sequence similarities between the α-domain of MATα1 and the HMG-boxes of many sequence specific DNA binding transcription factors have been recently studied and an evolutionary relationship between the α-domain and the HMG-box DNA binding motif has been proposed [13]. However, despite a wealth of structural information on the HMG-box structure to date with many experimental structures of HMG-box proteins available in the protein data bank, all the known HMG-box proteins currently available have an insignificant level of sequence identity to α-domain proteins.
The HMG-box proteins are crucial participants in many biological processes that involve chromosome architecture, and DNA metabolism (reviewed in [14,15]). Proteins that contain HMG-box domains usually fall within two general categories. The first category consists of non-sequence specific DNA binding proteins (HMGB-type) with two HMG-boxes followed by a long acidic C-tail. The second group consists of a diverse set of proteins (both sequence specific and non-sequence specific) with a single HMGbox domain without an acidic C-tail. The HMG-box domain consists mainly of three α-helices that pack in the form of an L-shaped molecule [16-31]. HMG-box domains interact primarily with the minor groove of DNA on the concave surface and cause significant distortions to the DNA helix. An example is illustrated in Figure 2. A basic N-terminal loop wraps around the DNA backbone and making specific contacts with the phosphates and bases. Sequence specific HMG-box proteins typically have a conserved Asn within the basic N-terminal extension that promotes a water mediated base interaction. The first helix will align within the minor groove with a significant curve. The first helix will insert hydrophobic residues (Met, Ile, Try, Val) between the stacked DNA bases. Although the majority of the DNA binding residues of HMG-boxes are found within the basic N-terminal extension and helix I, some DNA binding residues are found on the N-terminal side of Helix II [16,18,19,21,24-29]. In general, the interactions of the HMG-box with DNA leads to a highly distorted DNA structure with a wider minor groove and a significantly bent DNA helix. The DNA bending of HMG proteins facilitates the formation of higher-order nucleoprotein complexes necessary for transcription, recombination and DNA repair.
In the present study, the α-domain from the transcriptional activator MATα1 was chosen for modeling due to the sequence similarity and evolutionary relationship to the HMG-box proteins. In our modeling studies, we seek to identify the evolutionary conserved structural qualities that exist in both the α-domain and the HMG-box struc-
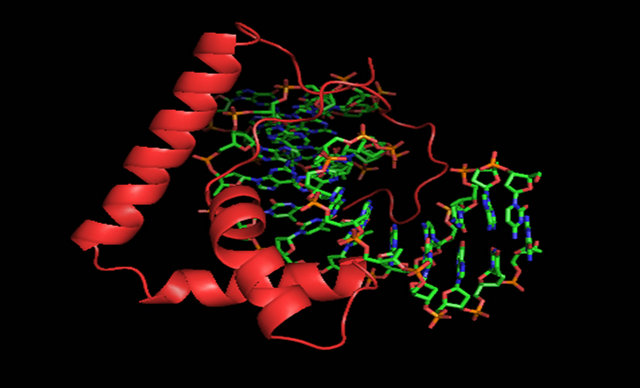
Figure 2. An example of an HMG-box protein. A complex of LEF-1 (cartoon, red) bound to DNA (stick, cpk, PDB ID 2LEF) [16].
ture. We also seek to examine parts of the α-domain that have diverged to give it its unique functions. A major difference between the α-domain and the HMG-box motif is the C-terminal 25 - 29 amino acids that do not contain an existing homology to any known structure.
2. Methods
2.1. MATα1 and α-Domain Target Sequence Determination
To find the MATα1 protein sequence and the α-domains target sequences for homology modeling, the full-length MATα1 from Saccharomyces cereviseae was retrieved from the National Center for Biotechnology Information (NCBI) database. This sequence was then used as a template in a BLAST (Basic-Local-Alignment-Search-Tool) search using NCBI-BLASTP suite [32]. The homologous domain between residues 81 and 146 was identified as the α-domain and used. The α-domains of organisms Ogataea angusta (CAE84418), Kluyveromyces lactis (Q08398), Lachancea waltii (CAO02575), Debaryomyces hansenii CBS767 (CAG88405), Zygosaccharomyces rouxii (CAR29078), Candida albicans SC5314 (EAK 95705) were determined by the same procedure and used for the sequence alignment.
2.2. Homology and ab Initio Modeling
To find the optimum HMG-box template for homology modeling, a search of the PDB (Protein Data Bank) was done for all structures containing an HMG-box domain. Initially, 42 PDB records were obtained however we omitted point mutants and highly homologous HMG-box structures to yield 27 representatives for comparisons (see Table A1). The HMG domain sequence files were taken directly from the PDB. Upon analyzing pairwise alignments using DIALIGN-TX [33], we obtained the best templates for homology modeling as PDB-ID: 2E6O (21.1%), 2LEF (14.2%). The final sequence alignment file of the target, template and the atomic coordinate file of the template structure was used to build the model using the SWISS-MODEL workspace. The corresponding model generated by the SWISS-MODEL workspace was subjected to multiple rounds of sidechain and loop adjustments and energy minimization procedures using the SWISS-PDB viewer [34] and the program GROMOS. For models containing the C-terminal domain we took the amino acids 81 - 175 and submitted it to the ROBETTA server [35,36]. After 5 - 6 weeks, the server found 5 possible structures for the domain. Each ROBETTA model and SWISS-MODEL was assessed as described below.
2.3. Model Assessment
The quality of the generated model was assessed by
Table 1. Structural models of the α-domain from Saccharomyces cereviseae.

checking the stereochemical parameters using PROCHECK [37], and ERRAT [38]. For each model we also checked its absolute quality by analyzing the QMEAN score, QMEAN Z-score, and the GA341 score [39-43]. The QMEAN is a scoring function consisting of a linear combination of six structural descriptors: 1) C-β interaction potential, 2) Solvation potential, 3) All-Atom interaction potential, 4) Tosional potential, 5) secondarystructure matching agreement (SSE-agree) and 6) solvent accessibility agreement (ACC-agree). The QMEAN score is a range from 0 to 1 with the most ideal value should be around 0.6 to 0.8. The QMEAN Z-score provides an estimate of the “degree of nativeness” of the structural model compared to experimental structures. High quality models will have a QMEAN Z-score less than 1 standard deviation from a similar sized high quality experimentally derived protein structure. The |QMEAN Z-scores| between 1 and 3 are acceptable quality and |QMEAN Z-scores| > 3 are considered “bad quality structures”. The GA341 is a score for the reliability of a model [40,41]. A model is predicted to be reliable when the model score is higher than a prespecified cutoff of 0.7. A reliable model has a probability of the correct fold that is larger than 95%.
2.4. Superposition and Merging
Superposition were done using the SWISS-PDB viewer [34] and the program SUPERPOSE [44]. RMSD values were computed in the program SUPERPOSE. Models containing bound DNA were prepared by superpositioning the model onto the structure of LEF-1/DNA using the Swiss-PDB viewer and omitting the LEF-1 structure from the picture.
3. Results
3.1. The Core of the α-Domain Contains an HMG-Box Fold
The similarities between the α-domain and the HMG-box have been established based on sequence identity [13]. However, despite extensive structural information on the HMG-box structure, no structure of the α-domain has been determined. Our motivation to do this work was to understand unique evolutionary aspects that exist between MATα1, the α-domain and the HMG-box containing proteins that are involved in mating-type determination. An initial BLAST search using the full-length MATα1, or the homologous α-domain did not reveal any sequence containing a solved structure in the PDB. Further analysis of the full-length MATα1 reveals a naturally disordered region (aa. 1 - 44) that exists on the N-terminus (data not shown). The C-terminus of MATα1 appears to form a structured core of which only residues 81 - 146 have sequence conservation in other yeast and HMG-box proteins. The region from amino acids 147 to 175 had only modest conservation in α-domain proteins of closely related yeasts. Secondary structural analysis of the C-terminal region of MATα1 from S. cereviseae reveals the presence of 5 or more helices (see Figure 3). The fifth helix was predicted with low confidence when analyzed using PsiPRED [45]. Other prediction gave similar results and thus the confidence scores for helix was slightly higher than for β-sheet and random coils. We then sought to compare the conserved region considered as the α-domain to all experimentally determined structures of HMG-box proteins. We were able to find 27 known structures of HMG-box domains that have been deposited into the PDB (see Table A1). All known structures were analyzed by a sequence structural alignment (data not shown). We took HMG-box domain sequences from the Protein Data Bank and α-domain sequences from the SWISSPROT database and performed a sequence alignment (Figures 4(a) and (b)). The sequence alignment was performed by the program MUSCLE and interestingly we found no difference in the alignment among the HMG-box proteins when only a sequencestructural alignment was performed. The critical amino acids within the HMG-box core are listed in Table A2. The α-domain protein from various ascomycete yeast were obtained and aligned along with the HMG-box proteins and colored in CLUSTALX [46] in JALVIEW [47] (see Figure 4 and Table A1). By comparing multiple models of structurally determined HMGbox domains, we were able to look very closely at common core elements among the HMG-box domains and determine how an α-domain would fold assuming the core of MATα1 folded as an HMG-box domain. Taken together, the HMG-box proteins shared very little sequence homology to the α-domain proteins with sequence homologies ranging from 5% to 21% identity (compared to the S. cerevisiae sequence). However, among the conserved sequence elements, the positions within the HMG-box that is required for either DNA binding or for hydrophobic interactions within the core are highly retained. A summary table is provided (see Table A2). Interestingly, residues involved in DNA binding include basic amino acids of the N-terminal loop, a conserved Asn at position 6 between the N-terminal loop and the first helix, and conserved intercalation at position 9 are conserved in all the α-domain proteins shown. The conserved Asn at position 6 is found in all sequence specific HMG-box proteins. The second intercalating residue of the first helix (at position 12) is an Arg in the α-domain which in the position to intercalate between the stacked bases, however, depending on the rotamer form used in the modeling, it can form a hydrogen bond to a nearby pyrimidine. Because Arg is a diversion away from a normal hydrophobic amino acid, this clearly represents a fundamental difference in how the α-domain binds to DNA relative to its close relatives of the HMG-box proteins. Other HMG-box domains have an Arg at this position, however, their structures have yet to be determined.
Despite high conservation of residues on the N-terminal loop and first half of helix 1, the rest of the sequence conservation is low. This exists when compareing all HMG-box proteins as well as α-domain proteins. However a close analysis reveals that amino acids that form hydrophobic contacts in the interior packing between the three helices are conserved. Aromatic residues at positions 8, 11, 42 and 53 are highly conserved in all HMG-box domains and present in all α-domains. Other conserved hydrophobic amino acids that stack between helix 1 and 2 are at positions 23 and 34. A diagram of these amino acids is in Figures 5(d) and (e).
 (a)
(a) (b)
(b)
Figure 3. Primary and secondary structural analysis of MATα1. (a) A domain chart of MATα1. (b) Secondary structural analysis of the CTD of MATα1. Confidence values were determined by PsiPRED [45]. Only Helix or Coil regions were found. Confidence values for Coil were given a negative sign and plotted in Microsoft EXCEL. The confidence scores for β-sheet were less than 2 and not considered significant except for regions 167 to 173 (data not shown).
 (a)
(a) (b)
(b)
Figure 4. Sequence alignment of selected α-domains and HMG-box domains. (a) The sequence alignment of selected α-domains from ascomycete yeasts with structurally determined HMG domains. Only Helix I, II and III of the HMG-box core (65 resudues) were aligned here. (b) The sequence alignment of the selected α-domain from ascomycete yeasts. Only Helix III, IV, and V are shown. All sequences were aligned using MUSCLE [48]. The abbreviations for the ascomycete yeasts are: a1_K: MATα1, Kluyveromyces lactis; a1_L: Lachancea waltii; a1_S: Saccharomyces cerevisiae; a1_Z: Zygosaccharomyces rouxii; a1_O: Ogataea angusta; a1_C: Candida albicans SC5314; a1_D: Debaryomyces hansenii CBS767. The HMG domains are listed by their PDB ID which is explained in the Table A1.
By using comparative homology modeling, we have developed homology models of the α-domain from a representative ascomycete yeast S. cerevisiae (see Table 1). The modeling of the α-domain was performed by two different homology modeling programs: Swiss-Model [34], and ROBETTA [35]. Using the SWISS-MODEL workspace, a manually aligned sequence/template with the α-domain (residues 81 - 146) and the HMG-box protein (PDB ID 2E6O) was used. The resulting model retained many of the features of the template but with few exceptions (see Figures 5(a)-(c)). First, the C-terminal end of helix 1 deviates significantly from the template model. The φ and ψ angles also deviate away from the α-helical structure. Second, helix 3 is only about twothirds the length of helix 3 in the template. The hydrophobic core of the models retained the packing arrangement of the aromatic residues at positions 8, 11, 42, and 53 (see Table A2, Figure 4(a), Figures 5(d) and (e)). When the models produced are superimposed onto LEF-1/DNA, the positions of the critical intercalating Met (position 9) are in very close proximity (see Figure 5(f)). The deviations from the template HMG-box structure is consistent among all known HMG-box proteins used as a comparison here (data not shown). These two deviated positions are also consistent with a previously determined homology model of the α-domain from pezizomycotan yeast using the PHYRE [13].
3.2. The C-Terminal Region Makes an Essential Contribution to the α-Domain
Using the SWISS-MODEL workspace, we could success-
 (a)
(a) (b)
(b) (c)
(c)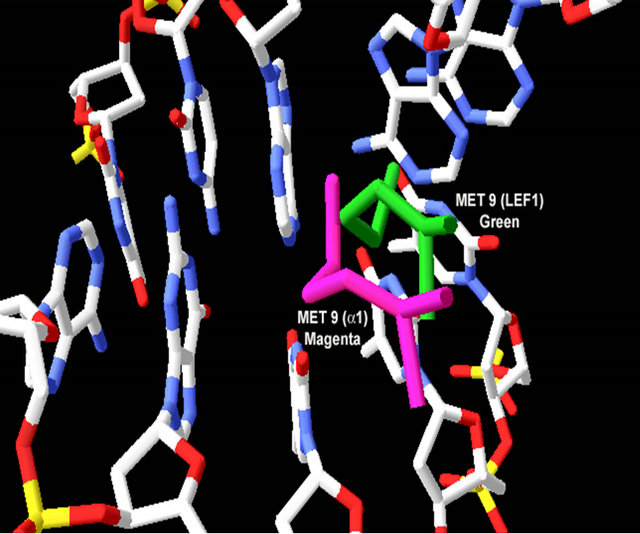 (d)
(d)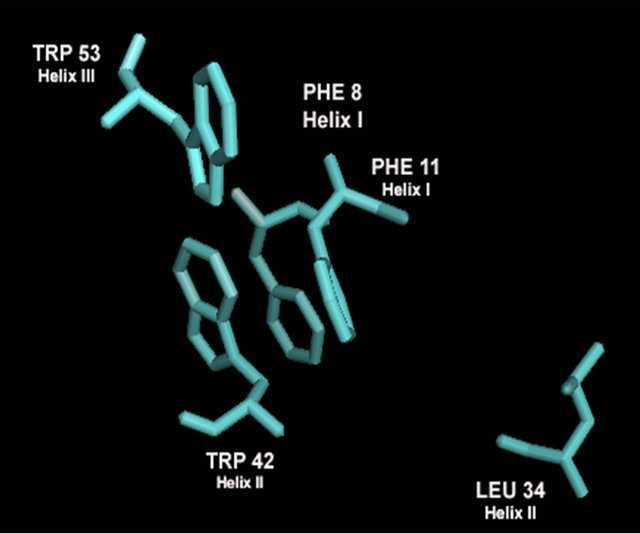 (e)
(e)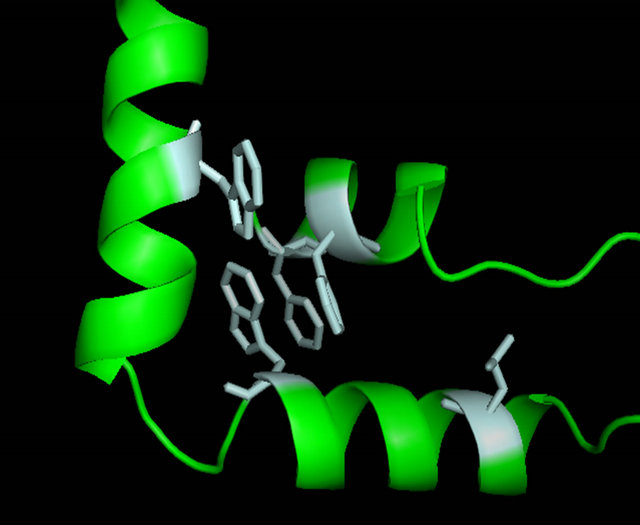 (f)
(f)
Figure 5. Evaluation of the homology model of α-domain (aa. 81 - 146) from Saccharomyces cereviseae. (a) An ribbon diagram of the homology modeled backbone (aa. 81 - 146) from the HMG-domain (PDB ID 2E6O). The model is shown as displayed in Swiss Model PDB viewer [34]. (b) an α-carbon trace of the superpositioned α-domain homology model (81 - 146) onto its template 2E6O. The model is in blue and the template is in red. (c) A superposition of the HMG core of 4 models (see table 2) onto the structure of LEF-1. Each model is shown in an α-carbon trace. The colors are: LEF-1 (Red), Model 1_SWM (Blue), Model 3_ROB (green), Model 4_ROB (yellow), Model 5_ROB (purple). (d) A diagram displaying the amino acids that form a hydrophobic core between Helix I, II and III in the modeled HMG-core part of Model_3_ROB are shown. (e) A diagram displaying the amino acids from “D” minus the backbone of the protein. (f) a diagram showing the intercalating amino acids of helix 1 (at position 9) of the superpositioned Model 3_ROB homology model (81 - 146) and LEF-1/DNA structure. In the display, only the positions of the amino acids at 9 are shown in the protein and only nearby DNA nucleotides are displayed. This model and Figures 5(b), (c) were generated in the Swiss PDB viewer. Figures 5(a), (d), and (e) were generated in PYMOL [49].
fully model the conserved region of the α-domain; however, analysis of the C-terminus indicated that this core region of the protein is not likely an independent domain. When modeled independently, several regions, particularly on helix 3, could not be repositioned without high residue error (data not shown). The amino acids at the C-terminus of helix 3 are concentrated with many hydrophobic groups which are not likely found exposed to solvent. We then used ROBETTA [35] to find a better structure that included the region of the C-terminus from amino acids 146 to 175. ROBETTA uses the ROSETTA [36] de novo and comparative modeling methods simultaneously to find full chain structural models. The de novo models are built through fragment insertion and simulated annealing. The whole 94 amino acid C-terminus (aa. 81 - 175) was determined by ROBETTA. ROBETTA returned five possible solutions to the structure of the C-terminal domain (see Table 1 and Figures 6(a)-(e)). Interestingly, couldn’t find any solutions for the 28 amino acid non-homologous C-terminal region (aa. 147 - 175) without the presence of the homologous HMGcore region (aa. 81 - 146) of the α-domain. The solutions to the HMG core that ROBETTA determined compared well with our HMG core using the SWISS-MODEL server and Swiss PDB viewer (see RMSD values in Table 1 and Figure 5(c)). The superposition of the modeled HMG core regions (81 - 146) from ROBETTA with the template 2E6O had a higher RMSD values than the superposition of the HMG core determined using the SWISSMODEL workspace and Swiss-PDB viewer (value of 1.71 Ǻ for Model 1_SWM verses an average of 2.46 Ǻ for Model 1 ROB through Model 5 ROB). The quality of the models was analyzed by the QMEAN, QMEAN Z-score [41-43] and the GA341 score from ModEval [39,40]. The QMEAN, QMEAN Z-score and GA341 are briefly discussed in the methods section. All models were analyzed by other structural analysis servers as well. The stereochemical analysis by PROCHECK [37] can be found in the Table A3. The results of the evaluation show that Model 3_ROB had the best quality scores when you compare the QMEAN score and the GA341 score of 1.000. The QMEAN score for Model 3_ROB is slightly lower than ideal range. We believe that this is due to its small size (94 amino acids) as compared to most of the test proteins analyzed. Analysis by PROCHECK reveals that Model 3_ROB has 85.4% of residues dihedral angles are in the most favored region (see Table A3). This is below the 90% for an ideal protein but much higher than the template used (2E6O) for modeling the HMG core region of Model 3_ROB. We also considered that a good model should be able to bind DNA stably and thus the C-terminus which has very little sequence conservation is likely not to participate in DNA binding. Each model was analyzed as to whether the C-terminus would interfere with DNA binding (see Table 1). The models were superimposed and merged onto the structure of LEF-1/DNA. The LEF-1 structure was then omitted and regions that clashed with DNA were highlighted. We did consider the possibility that minor conformational changes could occur upon DNA binding. Only Model 3_ROB, and Model 4_ROB could bind DNA reversibly without major conformational changes and all the amino acids that participate in DNA binding were available in these models. A picture of Model 3_ROB bound to DNA is in Figure 6(h). The C-terminus of Model 5_ROB does not interfere with DNA binding however; ROBETTA modeled a β-sheet in the N-terminal arm which would interfere with DNA binding.
When the C-terminal region of the α-domain (147 - 175 of S. cerevisiae) is analyzed among the ascomycete yeast species, only little conservation is observed (Figure 4(b)). A fourth helix appears with some conserved residues (aa. 69 - 73) “FVEWL”. A close look at Model 3_ROB reveals some elegant interactions between these conserved residues in the fourth helix. In S. cerevisiae, Phe 69, Trp 72 and Leu 73 of Helix IV form a hydrophobic pocket with Tyr 60 of Helix III (Figures 6(f) and 6(g)). Val 70 interacts with Helix I and the conserved acidic residue Glu 71 makes a contact to the basic N-terminal loop. These interactions are unique to the α-domain.
4. Discussion
The Zygomycota, which represents the early branch of the fungal evolutionary tree, contains an HMG-box domain in each of their two different mating-types. Detailed knowledge of the MAT loci of later divergent fungi shows the presence of a α-domain protein along with an HMG-box and homeodomain proteins. It would require only small changes in the DNA-binding domain to confer different promoter specificity required in divergent species of the ascomycota. It is possible that the acquisition of a unique DNA-binding domain by mutation of an existing HMG-box protein occurred concurrently with the evolution of the ascomycota MAT loci.
The evidence presented here as well should confirm that the α-domain belongs in the HMG-box superfamily. Our evidence for its placement is as follows: 1) conserva-
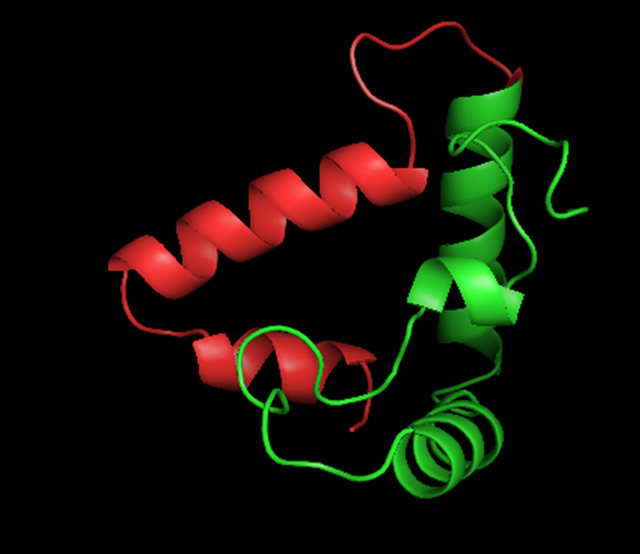 (a)
(a)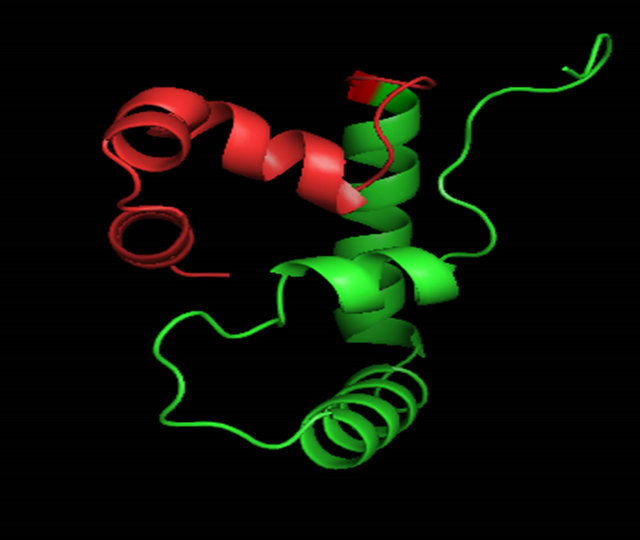 (b)
(b)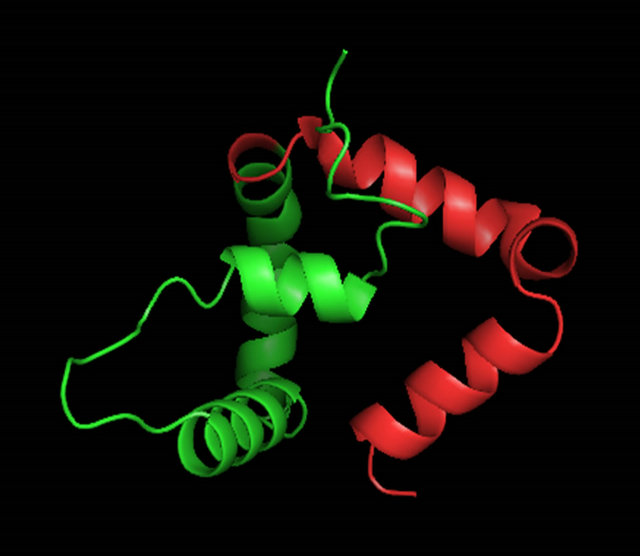 (c)
(c) (d)
(d)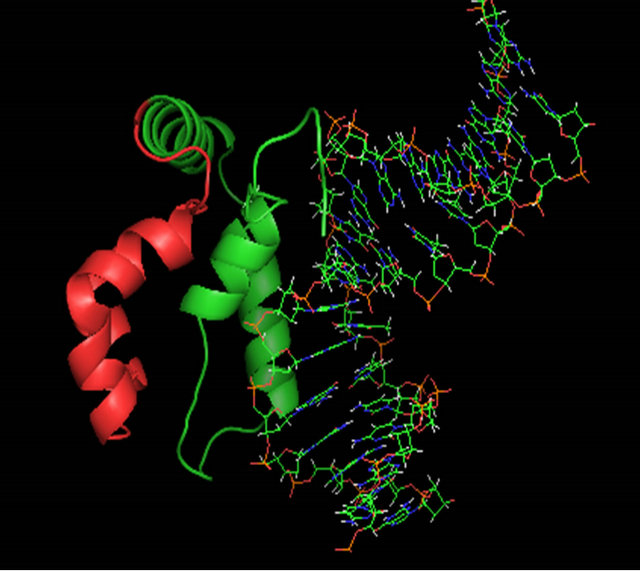 (e)
(e)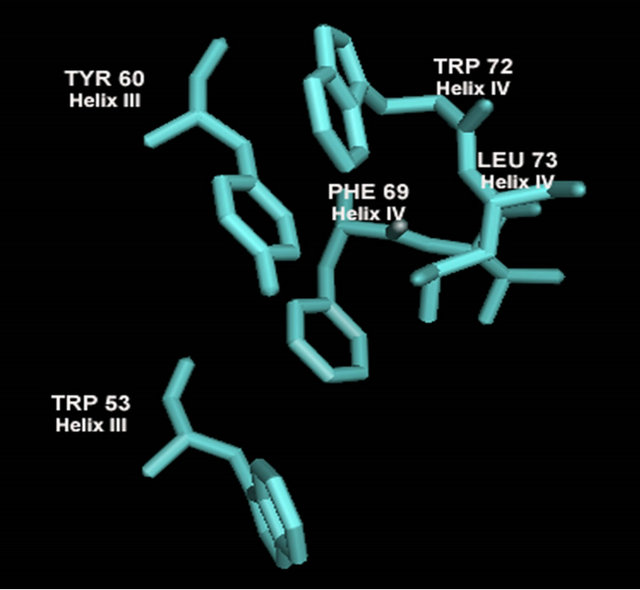 (f)
(f)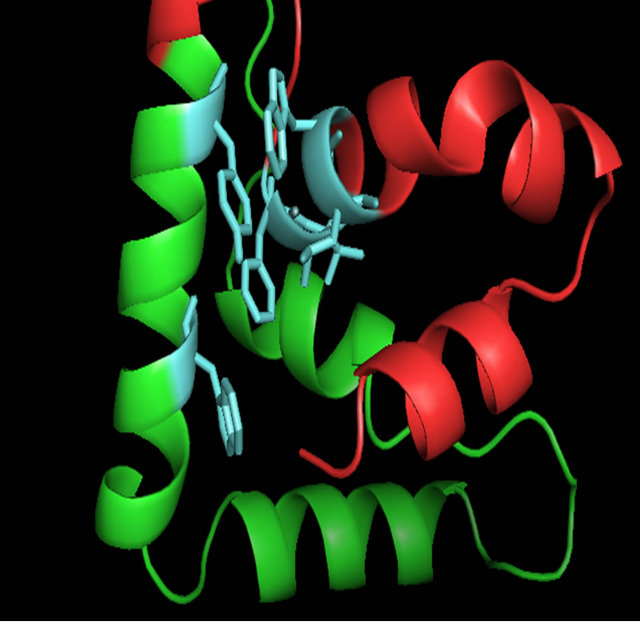 (g)
(g) (h)
(h)
Figure 6. A ribbon diagram of the C-terminus of MATα1 (aa. 81 - 175) determined by ROBETTA [24]. The HMG core is colored in green and the C-terminal extension (147 - 175) is colored in red. (a) Model 1_ROB. (b) Model 2_ROB. (c) Model 3_ROB. (d) Model 4_ROB. (e) Model 5_ROB. (f) Model 3_ROB with selected hydrophobic residues of Helix III and IV highlighted in torquiose. (g) Same as “F” except the ribbon backbone has been omitted. (h) Model 3_ROB positioned bound to DNA.
tion of crucial DNA binding residues at positions 6, 9, 12, and 31 (see Table A2); 2) Conservation of crucial hydrophobic residues required for an L-shaped tertiary structure characteristic of HMG-box proteins.
Despite strong conservation of the functional residues within the HMG-box core, the α-domain is very unique among HMG-box proteins. The major differences include a reduction in length of helix 1 and 3 and a nonhomologous C-terminal helical extension. The C-terminal extension will likely be different among major yeast species however some conservation in the fourth helix is observed. Our best model reveals a significant number of interactions of Helix IV with Helix III, Helix I and the N-terminal loop. The fourth Helix may have a role in regulation of DNA binding by stabilizing the unbound state of the HMG-box domain.
Here we demonstrate that in S. cereviseae, the C-terminal extension contains a three helical extension that contacts Helix I, Helix III and the N-terminal loop of the HMG-box core. The question remains as to how the diverging sequence and structure lead to a diverging function of MATα1. One interesting aspect of MATα1, is that DNA binding in vivo is believed to occur only in the presence of MCM1. The binding to QP’ DNA by MATα1 alone in vitro is very weak (unpublished work D. Jackson, S. Tan) and requires the assistance of MCM1. This reduced or regulated binding by MATα1 has yet to be explored in detail.
Here we are able to present a structural model of MATα1 (Best model: Model 3_ROB, Figure 6(c)) and how this protein binds to DNA. MATα1 has direct protein protein interactions to MCM1 and possibly STE12 [7,10,11]. The best characterized is its interaction with MCM1 [10]. The structural model presented here will be a valuable asset in our understanding of how the MATα1/MCM1/DNA complex activates transcription of α-specific genes which is an ancient outstanding model for how gene expression occurs. Several colleagues have found it difficult to crystallize MATα1, the α-domain or the MATα1/MCM1/DNA complex due to limited solubility and aggregation problems (D. Jackson, unpublished). The present model suggests that a soluble domain exists bound to DNA using the C-terminal fragment from residues 81 - 175. This is consistent with our experimental results on the solubility of MATα1 constructs (to be published elsewhere). Our model of the C-terminus is currently being used to design new constructs for crystallization and X-ray diffraction analysis. We hope to be reporting the experimentally determined 3-D structure of the α-domain or the structure of MATα1 soon.
5. Acknowledgements
We would like to thank Dr. Song Tan of the Department of Biochemistry and Molecular Biology at Pennsylvania State University for his preliminary work on MATα1 and his support of Dr. Doba Jackson from 2002-2006. We would also like to thank Dr. Maureen Murphy at Huntingdon College for helping to support Dr. Doba Jackson and an undergrad student Tarnisha Lawson. Finally, we would like to thank the National Science Foundation for a post-doctoral fellowship (award #-0310267) to Dr. Doba Jackson for years 2003-2006.
REFERENCES
- L. A. Casselton, “Fungal Sex Genes—Searching for the Ancestors,” BioEssays, Vol. 30, No. 8, 2008, pp. 711-714. doi:10.1002/bies.20782
- G. F. Sprague, J. Rine and I. Herskowitz, “Control of Yeast Cell Type by the Mating-Type Locus: II. Genetic Interactions between MATα and Unlinked α-Specific STE Genes,” Journal of Molecular Biology, Vol. 153, No. 2, 1981, pp. 323-335. doi:10.1016/0022-2836(81)9.0281-3
- S. Lee, N. Corradi, S. Doan, F. S. Dietrich, P. J. Keeling and P. J. Keeling, “Evolution of the Sex-Related Locus and Genomic Features Shared in Microsporidia and Fungi,” PLos One 5, 2010, Article ID: 10539. doi:10.1371/journal.pone.0010539
- G. F. Sprague, Jr, L. C. Blair and J. Thorner, “Cell Interactions and the Regulation of Cell Type in Yeast Saccharomyces cerevisiae,” Annual Review of Microbiology, Vol. 37, 1983, pp. 623-660. doi:10.1146/annurev.mi.37.100183.003203
- D. R. Scannell, G. Butler and K. H. Wolfe, “Yeast Genome Evolution: The Origin of the Species,” Yeast, Vol. 24, No. 11, 2007, pp. 929-942.
- T. Koestler and I. Ebersberger, “Zygomycetes, Microsporidia, and the Evolutionary Ancestry of Sex Determination,” Genome Biology and Evolution, Vol. 3, 2011, pp. 186-194. doi:10.1093/gbe/evr009
- D. C. Hagan, L. Bruhn, C. Westby and G. F. Sprague, “Transcription of α-Specific Genes in Saccharomyces cerevisiae: DNA Sequence Requirements for Activity of the Coregulator Alpha1,” Molecular and Cellular Biology, Vol. 13, No. 11, 1993, pp. 6866-6875.
- P. Shore and A. Sharrocks, “The MADS-Box Family of Transcription Factors,” European Journal of Biochemistry, Vol. 229, 1995, pp. 1-13. doi:10.1111/j.1432-1033.1995.tb20430.x
- S. Tan and T. Richmond, “Crystal Structure of the Yeast MATα2/MCM1/DNA Ternary Complex,” Nature, Vol. 391, No. 6668, 1998, pp. 660-666. doi:10.1038/35563
- S. Tan, G. Ammerer and T. Richmond, “Interactions of Purified Transcription Factors: Binding of Yeast MATα1 and PRTF to Cell Type Specific, Upstream Activating Sequences,” EMBO Journal, Vol. 7, No. 13, 1988, pp. 4255-4264.
- E. Carr, J. Mead and A. Vershon, “α1-Induced DNA Bending Is Required for Transcriptional Activation by the MCM1-α1 Complex,” Nucleic Acid Research, Vol. 32, No. 8, 2004, pp. 2298-2305. doi:10.1093/nar/gkh560
- F. Lim, A. Hayes, A. West, A. Pic-Taylor, Z. Darieva, et al., “Mcm1p-Induced DNA Bending Regulates the Formation of Ternary Transcription Factor Complexes,” Molecular and Cellular Biology, Vol. 23, No. 2, 2003, pp. 450-461. doi:10.1128/MCB.23.2.450-461.2003
- T. Martin, S.-W. Lu, H. Tilbeurgh, D. R. Ripoll, C. Dixelius, et al., “Tracing the Origin of the Fungal α1-Domain Places Its Ancestor in the HMG-Box Superfamily: Implication for Fungal Mating-Type Evolution,” PLos One 5: 2010, Article ID: e15199. doi:10.1371/journal.pone.0015199
- R. Reeves, “HMG Nuclear Proteins: Linking Chromatin Structure to Cellular Phenotype,” Biochim Biophys Acta, Vol. 1799, No. 1-2, 2010, p. 3.
- M. Stros, D. Launholt and K. Grasser, “The HMG-Box: A Versatile Protein Domain Occuring in a Wide Variety of DNA-Binding Proteins,” Cellular and Molecular Life Sciences, Vol. 64, No. 19-20, 2007, pp. 2590-2606. doi:10.1007/s00018-007-7162-3
- J. J. Love, X. Li, D. A. Case, K. Giese, R. Grosschedl and P. E. Wright, “Structural Basis for DNA Bending by the Architectural Transcription Factor LEF-1,” Nature, Vol. 376, No. 6543, 1995, pp. 791-795. doi:10.1038/376791a0
- H. Rhong, Y. Li, X. Shi, X. Zhang, Y. Gao, H. Dai, M. Teng, L. Niu, Q. Liu and Q. Hao, “Structure of Human Upstream Binding Factor HMG-Box 5 and Site for Binding of the Cell-Cycle Regulatory Factor TAF1,” Acta Crystallography Section D, Vol. D63, 2007, pp. 730-737. doi:10.1107/S0907444907017027
- F. V. Murphy 4th, R. M. Sweet and M. E. Churchill, “The Structure of a Chromosomal High Mobility Group Protein-DNA Complex Reveals Sequence-Neutral Mechanisms Important for Non-Sequence-Specific DNA Recognition,” EMBO Journal, Vol. 18, No. 23, 1999, pp. 6610- 6618. doi:10.1093/emboj/18.23.6610
- U. M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, “Basis for Recognition of Cisplatin-Modified DNA by High-Mobility-Group Proteins,” Nature, Vol. 399, No. 6737, 1999, pp. 708-712. doi:10.1038/21460
- H. M. Weir, P. J. Kraulis, C. S. Hill, A. R. Raine, E. D. Laue and J. O. Thomas, “Structure of the HMG-Box Motif in the β-Domain of HMG1,” EMBO Journal, Vol. 12, No. 4,1993, pp. 1311-1319.
- C. H. Hardman, R. W. B. Hurst, A. R. Raine, K. D. Grasser, J. O. Thomas and E. D. Laue, “Structure of the A-Domain of HMG1 and Its Interaction with DNA as Studied by Heteronuclear Threeand Four-Dimensional NMR Spectroscopy,” Biochemistry, Vol. 34, No. 51, 1995, pp. 16596-16607. doi:10.1021/bi00051a007
- A. Remenyi, K. Lins, L. J. Nissen and R. Reinbold, “Crystal Structure of the Pou/HMG/DNA Ternary Complex Suggests Differential Assembly of Oct4 and Sox2 on Two Enhancers,” Genes & Development, Vol. 17, No. 18, 2003, pp. 2048-2059.
- Y. Xu, W. Yang, J. Wu and Y. Shi, “Solution Structure of the First HMG-Box Domain in Human Upstream Binding Factor,” Biochemistry, Vol. 41, No. 17, 2002, pp. 5415- 5420. doi:10.1021/bi015977a
- M. H. Werner, J. R. Huth, A. M. Gronenborn and G. M. Clore, “Molecular Basis of Human 46X,Y Sex Reversal Revealed from the Three-Dimensional Solution Structure of the Human SRY-DNA Complex,” Cell, Vol. 81, No. 5, 1995, pp. 704-705.
- P. Palasingam, R. Jauch, C. K. L. Ng and P. R. Kolatkar, “The Structure of Sox17 Bound to DNA Reveals a Conserved Bending Topology but Selective Protein Interaction Platforms,” Journal of Molecular Biology, Vol. 388, No. 3, 2009, pp. 619-630.
- T. A. Gangelhoff, P. S. Mungalachetty, J. C. Nix and M. E. Churchill, “Structural Analysis and DNA Binding of the HMG Domains of the Human Mitochondrial Transcription Factor A,” Nucleic Acids Research, Vol. 37, No. 10, 2009, pp. 3153-3164. doi:10.1093/nar/gkp157
- K. Stott, G. S. Tang, K. B. Lee and J. O. Thomas, “Structure of Tandem HMG-Boxes and DNA,” Journal of Molecular Biology, Vol. 360, No. 1, 2006, pp. 90-104. doi:10.1016/j.jmb.2006.04.059
- D. C. Williams, M. Cai and G. M. Clore, “Molecular Basis for Synergistic Transcriptional Activation by Oct1 and Sox2 Revealed from the Solution Structure of the 42-kDa Oct1·Sox2·Hoxb1-DNA Ternary Transcription Factor Complex,” The Journal of Biological Chemistry, Vol. 279, No. 2, 2004, pp. 1449-1457. doi:10.1074/jbc.M309790200
- J. E. Masse, B. Wong, Y.-M. Yen, F. H.-T. Allain, R. C. Johnson and J. Feigon, “The S. cerevisiae Architectural HMGB Protein NHP6A Complexed with DNA: DNA and Protein Conformational Changes upon Binding,” Journal of Molecular Biology, Vol. 323, No. 2, 2002, pp. 263-284. doi:10.1016/S0022-2836(02)00938-5
- P. D. Cary, C. M. Read, B. Davis, P. C. Driscoll and C. Crane-Robinson, “Solution Structure and Backbone Dynamics of the DNA-Binding Domain of Mouse Sox-5,” Protein Science, Vol. 10, No. 1, 2001, pp. 83-98. doi:10.1110/ps.32801
- N. Kasai, Y. Tsunaka, I. Ohki, S. Hirose, K. Morikawa and S. Tate, “Solution Structure of the HMG-Box Domain in the SSRP1 Subunit of FACT,” Journal of Biomolecular NMR, Vol. 32, No. 1, 2005, pp. 83-88. doi:10.1007/s10858-005-3662-3
- C. Camacho, G. Coulouris, V. Avagyan N. Ma, J. Papadopoulos, et al., “BLAST+: Architecture and Applications,” BMC Bioinformatics, Vol. 10, 2008, p. 421. doi:10.1186/1471-2105-10-421
- S. Amarendran, J. W. Menkhoff, M. Kaufmann and B. Morgenstern, “DIALIGN-TX: An Improved Algorithm for Segment Based Multiple Sequence Alignment,” BMC Bioinformatics, Vol. 6, 2005, p. 66. doi:10.1186/1471-2105-6-66
- N. Guex and M. C. Peitsch, “SWISS-MODEL and the Swiss Pdb Viewer: An Environment for Comparative Protein Modeling,” Electrophoresis, Vol. 18, No. 15, 1997, pp. 2714-2723. doi:10.1002/elps.1150181505
- D. E. Kim, D. Chivian and D. Baker, “Protein Structure Prediction and Analysis Using the Robetta Server,” Nucleic Acids Research, Vol. 32, No. 2, 2004, pp. W526- W531. doi:10.1093/nar/gkh468
- S. Raman, R. Vernon, J. Thompson, M. Tyka, R. Sadreyev, J. Pei, D. Kim, E. Kellogg, F. DiMaio, O. Lange, L. Kinch, W. Sheffler, B. Kim, R. Das, N. V. Grishin and D. Baker, “Structure Prediction for CASP8 with All-Atom Refinement Using Rosetta,” Proteins: Structure, Function and Bioinformatics, Vol. 77, No. S9, 2009, pp. 89-99. doi:10.1002/prot.22540
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, “PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures,” Journal of Applied Crystallography, Vol. 26, 1993, pp. 283-291. doi:10.1107/S0021889892009944
- C. Colovos and T. O. Yeates, “Verifications of Protein Structures: Patterns of Nonbonded Atomic Interactions.” Protein Science, Vol. 2, No. 9, 1993, pp. 1511-1519. doi:10.1002/pro.5560020916
- F. Melo, R. Sanchez and A. Sali, “Statistical Potentials for Fold Assessment,” Protein Science, Vol. 11, No. 2, 2002, pp. 430-448.
- D. Eramian, N. Eswar, M. Y. Shen and A. Sali, “How Well Can the Accuracy of Comparative Protein Structure Models Be Predicted?” Protein Science, Vol. 17, No. 11, 2008, pp. 1881-1893. doi:10.1110/ps.036061.108
- P. Benkert, S. C. E. Tosatto and D. Schomburg, “QMEAN: A Comprehensive Scoring Function for Model Quality Assessment,” Proteins: Structure, Function, and Bioinformatics, Vol. 71, No. 1, 2008, pp. 261-277. doi:10.1002/prot.21715
- P. Benkert, M. Biasini and T. Schwede, “Toward the Estimation of the Absolute Quality of Individual Protein Structure Models,” Bioinformatics, Vol. 27, No. 3, 2010, pp. 343-350. doi:10.1093/bioinformatics/btq662
- P. Benkert, M. Künzli and T. Schwede, “QMEAN Server for Protein Model Quality Estimation,” Nucleic Acids Research, Vol. 37, 2009, pp. 510-540. doi:10.1093/nar/gkp322
- E. Krissinel and K. Henrick, “Secondary-Structure Matching (SSM), a New Tool for Fast Protein Structure Alignment in Three Dimensions,” Acta Crystallographica Section D, Vol. 60, No. 1, 2004, pp. 2256-2268. doi:10.1107/S0907444904026460
- L. J. Mcgruffin, K. Bryson and D. T. Jones, “The PSIPRED Protein Structure Prediction Server,” Bioinformatics, Vol. 16, No. 4, 2000, pp. 404-405.
- J. D. Thompson, T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, “The ClustalX Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools,” Nucleic Acids Research, Vol. 24, 1997, pp. 4876-4882.
- A. M. Waterhouse, J. B. Procter, D. M. A. Martin, M. Clamp and G. J. Barton, “Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench,” Bioinformatics, Vol. 25, No. 9, 2009, pp. 1189-1191. doi:10.1093/bioinformatics/btp033
- R. C. Edgar, “MUSCLE, Multiple Sequence Alignment with High Accuracy and High Thoroughput,” Nucleic Acid Research, Vol. 32, No. 5, 2004, p. 1792.
- “The PyMOL Molecular Graphics System,” Version 1.5.0.4, Schrödinger, 2006.
Appendix
Table A1. Structural comparison of HMG-box domains.
| PDB ID | Protein | Organism | Method | Ref. |
| 2HDZ | UBF HMG #5 | Hom Sap | X-Ray | [17] |
| 1J3C | C-terminal domain of HMGB2 | Sus scrofa | NMR | NP |
| 1QRV | HMG-D/DNA complex | Dros. mel. | X-Ray | [18] |
| 2LEF | Transcription factor LEF1/DNA complex | Mus mus | NMR | [16] |
| 1CKT | HMG #1 bound to cisplatin-DNA | Rat Norv | X-Ray | [19] |
| 1J3X | N-terminal domain of HMGB2 | Sus scrofa | NMR | NP |
| 1HMF | β-domain of HMG1 | Rat Norv | NMR | [20] |
| 2YUL | HMG-box of SOX-17 | Hom Sap | NMR | NP |
| 2YQI | Second HMG domain of HMGB3 | Hom Sap | NMR | NP |
| 2EQZ | First HMG domain of HMGB3 | Hom Sap | NMR | NP |
| 1V63 | UBF1 #6 | Mus mus | NMR | NP |
| 1AAB | α-domain of HMG1 | Rat Norv | NMR | [21] |
| 1GTO | Oct4/SOX2/DNA complex | Mus mus | X-Ray | [22] |
| 1V64 | UBF1 #3 | Mus mus | NMR | NP |
| 1K99 | UBF1 #1 | Hom Sap | NMR | [23] |
| 1HRY | SRY/DNA | Hom Sap | NMR | [24] |
| 1WGF | UBF1 #4 | Mus mus | NMR | NP |
| 3F27 | SOX-17/DNA complex | Mus mus | X-Ray | [25] |
| 3FGH | Mitochondrial TF-A | Hom Sap | X-Ray | [26] |
| 2YUK | Myeloid Lymphoid Leukemia protein 3 | Hom Sap | NMR | NP |
| 2D7L | HMG-box/WD repeat protein | Hom Sap | NMR | NP |
| 2E6O | HMG-box transcription factor 1 | Hom Sap | NMR | NP |
| 2GZK | β-domain of HMGB1 | Rat Norv | X-Ray | [27] |
| 2CTO | Hypothetical protein FLJ14904 | Hom Sap | NMR | NP |
| 2CRJ | HMG domain protein HMGX2 | Mus mus | NMR | NP |
| 2CS1 | DNA mismatch repair protein | Hom Sap | NMR | NP |
| 2CO9 | Thymus HMG protein | Mus mus | NMR | NP |
| 1WZ6 | Bobby Sox homologue | Mus mus | NMR | NP |
| 1O4X | Oct1/SOX-2/DNA complex | Hom Sap | NMR | [28] |
| 1J5N | NHP6A complexed to DNA | S. cerev | NMR | [29] |
| 1I11 | SOX-5 | Mus mus | NMR | [30] |
| 1HSM | HMG protein (Hamster) | Cric gris | NMR | NP |
| 1WXL | SSRP subunit of FACT | Dros. mel. | NMR | [31] |
Table A2. Conserved positions in HMG-box proteins and the α-domain.
| Position* | AA-type | Function | HMG-box | α-domain |
| 2 - 4 | Basic (K,R) | makes critical backbone contacts to the DNA minor groove | +++ | +++ |
| 6 | S, N | makes water mediated contacts to bases/sugar residues in the minor groove | +++ | +++ |
| 8, 11 | Aromatic (F,Y,W) | Positions 8 and 11 interacts with the hydrophobic core | +++ | +++ |
| 9 | Hydrophobic (M,I,L) | intercalates between stacked bases | +++ | +++ |
| 12 | Any | intercalates between stacked bases in HMG domains | ++ | ?? |
| 14 | Acidic (D,E) | makes contacts with helix 2 | + | −− |
| 16 | Basic (R) | makes critical backbone contacts | ++ | −− |
| 23 | Hydrophobic (L,I) | makes hydrophobic contacts to helix 2 | +++ | +++ |
| 25 | P | defines the loop 1 region | +++ | −/+ |
| 33 | Acidic (D,E) | Unknown | +++ | +++ |
| 34 | Hydrophobic (L,I,V,A) | Hydrophobic contacts with helix 1 | +++ | +++ |
| 38 | S or T | Unknown | + | +++ |
| 42 | Hydrophobic (W) | makes hydrophobic contacts to helix 1 | +++ | +++ |
| 49 | Acidic (D,E) | Unknown | +++ | −− |
| 50 | Basic (K) | Unknown | +++ | +/− |
| 53 | Aromatic (W) | makes hydrophobic contacts to helix 1 | +++ | +++ |
| 57 | Hydrophobic (A) | makes contacts to the unstructured loop 1 | +++ | +++ |
*Position numbers correspond to the numbers from the scale at the top of Figure 4(a). Thus position 1 is actually position 89 in the S. cerevisiae protein sequence.
Table A3. PROCHECK results of structural models.
| Model name | Residues | Most favorable region | Additional allowed region | Generous allowed region | Disallowed region |
| SWM_1_Model | 81 - 146 | 85.7 | 14.3 | 0.8 | 0 |
| Rob_1_Model | 81 - 175 | 91.5 | 8.5 | 0 | 0 |
| Rob_2_Model | 81 - 175 | 91.5 | 8.5 | 0 | 0 |
| Rob_3_Model | 81 - 175 | 85.4 | 14.6 | 0 | 0 |
| Rob_4_Model | 81 - 175 | 87.8 | 12.2 | 0 | 0 |
| Rob_5_Model | 81 - 175 | 86.6 | 13.4 | 0 | 0 |
| 2E6O | --- | 76.3 | 23.7 | 0 | 0 |