Computational Chemistry
Vol.05 No.02(2017), Article ID:75774,9 pages
10.4236/cc.2017.52006
Comparative Study of the Chemical Reactivity of Helical Peptide Models for Protein Glycation
Juan Frau1, Daniel Glossman-Mitnik1,2
1Departament de Química, Universitat de les Illes Balears, Palma de Mallorca, Spain
2Laboratorio Virtual NANOCOSMOS, Departamento de Medio Ambiente y Energía, Centro de Investigación en Materiales Avanzados, Chihuahua, Mexico

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 4, 2017; Accepted: April 25, 2017; Published: April 28, 2017
ABSTRACT
Non-enzymatic glycation of proteins has been implicated as an important cause of the complications associated with diabetes and Alzheimer disease. It is well known that glycation involves the reactivity of, primarily, the  -amino group of the lysines present in the protein. The immediate chemical envi- ronment of an amino group modulates the glycation reaction. In this work, several model helical peptides for protein glycation has been studied by resorting to QM:MM calculations through the ONIOM methodology. Some Conceptual DFT descriptors have been calculated that allowed the com- parison of the chemical reactivity between the different model peptides in terms of the position of the Lys group and other spatially proximate amino acid residues.
-amino group of the lysines present in the protein. The immediate chemical envi- ronment of an amino group modulates the glycation reaction. In this work, several model helical peptides for protein glycation has been studied by resorting to QM:MM calculations through the ONIOM methodology. Some Conceptual DFT descriptors have been calculated that allowed the com- parison of the chemical reactivity between the different model peptides in terms of the position of the Lys group and other spatially proximate amino acid residues.
Keywords:
Helical Model peptides, Protein Glycation, Computational Chemistry, Molecular Modeling, Conceptual DFT, Chemical Reactivity Theory

1. Introduction
The nonenzymatic reaction between reducing carbohydrates and the amino groups of amino acids, peptide and proteins is called glycation and through a series of chain reaction leads to the formation of Advanced Glycation End- products (AGEs). These molecules have been related to some diseases like diabetes, Alzheimer and Parkinson. Nonenzymatic glycation of proteins has been implicated as an important cause of the complications associated with diabetes and Alzheimer disease. It is well known that glycation involves the reactivity of, primarily, the ε-amino group of the lysines present in the protein.
The immediate chemical environment of an amino group modulates the glycation reaction. Some years ago, Venkatraman et al [1] studied several helical peptide models in order to elucidate the proximity effect in the catalysis of the Amadori rearrangement. Their conclusions were similar to those of Howard et al [2] and Povey et al [3] who found that the glycation reaction affected the helicity, charge and other properties of the resulting molecules.
Therefore, we consider that it will be of practical interest to develop a the- oretical and computational tool that could be an aid for the prediction of the preferred glycation sites for the different possible conformational structures of model peptides because they could be an aid in the development of AGE in- hibitors.
Thus, the objective of this work is to predict the preferred glycation sites of the model helical peptides proposed by Venkatraman et al [1] by resorting to the calculation of some Conceptual DFT descriptors like the Fukui function indexes, the condensed dual descriptor 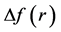 and the electrophilic and nucleophilic Parr functions. As the preferred glycation sites will be the same as those for the protonation reaction, the results of the calculations will allow us to qualitatively predict the pKa’s of the different lysine residues on the light of the obtained values for the Conceptual DFT descriptors [4] [5] [6] .
and the electrophilic and nucleophilic Parr functions. As the preferred glycation sites will be the same as those for the protonation reaction, the results of the calculations will allow us to qualitatively predict the pKa’s of the different lysine residues on the light of the obtained values for the Conceptual DFT descriptors [4] [5] [6] .
2. Theoretical Background
As this work is part of an ongoing project, the theoretical background is analog to that presented in previous research [7] - [13] and will be shown here for the sake of completeness. As it has been described in detail before, [7] - [13] within the conceptual framework of DFT, [5] [14] the chemical potential m is defined as: 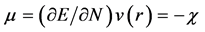 where
where 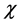 is the electronegativity, while the global chemical hardness
is the electronegativity, while the global chemical hardness  is:
is: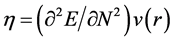 . Using a finite difference approximation, these expressions can be written as:
. Using a finite difference approximation, these expressions can be written as: 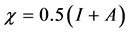 and
and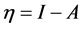 , where
, where 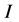 and
and 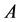 are the vertical ionization potential and elec- tron affinity, respectively. The electrophilicity index
are the vertical ionization potential and elec- tron affinity, respectively. The electrophilicity index  has been defined as:
has been defined as: . The electrodonating (
. The electrodonating (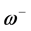 ) and electroaccepting (
) and electroaccepting (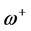 ) powers have been defined as:
) powers have been defined as:  and
and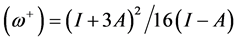 . It follows that a larger (
. It follows that a larger ( ) value corresponds to a better capability of accepting charge, whereas a smaller value of (
) value corresponds to a better capability of accepting charge, whereas a smaller value of ( ) makes it a better electron donor. In order to compare (
) makes it a better electron donor. In order to compare (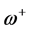 ) with (
) with (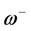 ), the following definition of net electrophilicity has been proposed:
), the following definition of net electrophilicity has been proposed: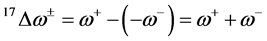 , that is, the electroaccepting power relative to the electrodonating power.
, that is, the electroaccepting power relative to the electrodonating power.
3. Settings and Computational Methods
Following the lines of our previous work [7] - [13] , the molecular structures of the studied compounds were constructed by starting with the readily available MOL structures for the amino acids (ChemSpider: www.chemspider.com, PubChem: https://pubchem.ncbi.nlm.nih.gov/). The most stable conformers were obtained by means of the Avogadro 1.2.0 program [18] [19] through a random sampling with Molecular Mechanics techniques and a consideration of all the torsional angles through the general AMBER force field [20] .
The energies of the neutral, positive and negative peptides were obtained within the framework of QM:MM calculations performed through the ONIOM method [21] in the presence of water as a solvent, by doing IEF-PCM computations according to the SMD solvation model [22] . For the QM region, the MN12SX density functional [23] in connection with the Def2TZVP basis set was chosen [24] [25] . The MN12SX density functional is a range-separated hybrid nonse- parable meta-NGA [23] . The Amber force field [26] was considered for the MM part. All the ONIOM calculations were performed as implemented in the Gaussian 09 [27] series of programs.
4. Results and Discussion
The following peptides proposed by Venkatraman et al [1] were studied as mentioned before:
KD4: Ac-EYUALKAUADUAAUR-NH2
KD2: Ac-EYUALUAKADUAAUR-NH2
KH4: Ac-EYUALKAUAHUAAUR-NH2
RKD4: Ac-EYRULKAUADUAAUA-NH2
K14: Ac-EYUALKAUAUAAUR-NH2
From the cluster analysis, some members were selected for further analysis: 3 for KD4, 4 for KD2, 3 for KH4, 3 for RKD4 and 1 for K14.
The ionization potentials 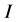 and electron affinities
and electron affinities 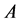 (in eV), and global electronegativity
(in eV), and global electronegativity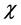 , total chemical hardness
, total chemical hardness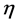 , global electrophilicity
, global electrophilicity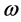 , electrodonating power, (
, electrodonating power, (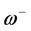 ), electroaccepting power (
), electroaccepting power (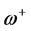 ), and net electrophilicity
), and net electrophilicity 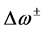 of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/ Def2TZVP:Amber) using water as solvent simulated with the SMD parame- trization of the IEF-PCM model are presented in Table 1.
of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/ Def2TZVP:Amber) using water as solvent simulated with the SMD parame- trization of the IEF-PCM model are presented in Table 1.
Within Conceptual DFT, the Fukui function is defined in terms of the derivative of 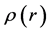 with respect to N [5] :
with respect to N [5] : 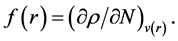 The function
The function 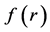 reflects the ability of a molecular site to accept or donate electrons. High values of
reflects the ability of a molecular site to accept or donate electrons. High values of 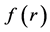 are related to a high reactivity at point r [5] .
are related to a high reactivity at point r [5] .
By applying a finite difference approximation to the previous expression, two definitions of Fukui functions depending on total electronic densities are obtained: 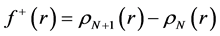 and
and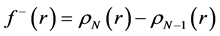 , where
, where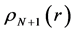 ,
, 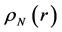 and
and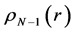 , are the electronic densities at point r for the system with
, are the electronic densities at point r for the system with ,
,  and
and 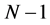 electrons, respectively. The first one,
electrons, respectively. The first one, 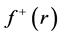 , has been associated to reactivity for a nucleophilic attack so that it measures the intra- molecular reactivity at the site r towards a nucleophilic reagent. The second one,
, has been associated to reactivity for a nucleophilic attack so that it measures the intra- molecular reactivity at the site r towards a nucleophilic reagent. The second one, 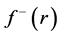 , has been associated to reactivity for an electrophilic attack so that this function measures the intramolecular reactivity at the site r toward an electrophilic reagent [14] .
, has been associated to reactivity for an electrophilic attack so that this function measures the intramolecular reactivity at the site r toward an electrophilic reagent [14] .
Morell et al. [28] - [34] have proposed a local reactivity descriptor (LRD) which is called the dual descriptor (DD) 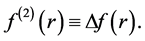 The dual descriptor can be condensed over the atomic sites: when
The dual descriptor can be condensed over the atomic sites: when 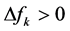 the process is driven by a
the process is driven by a

Table 1. The ionization potentials I and electron affinities A (in eV), and global elec- tronegativity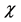 , otal chemical hardness
, otal chemical hardness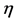 , global electrophilicity
, global electrophilicity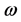 , electrodonating power, (
, electrodonating power, (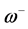 ), electroaccepting power (
), electroaccepting power (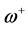 ), and net electrophilicity
), and net electrophilicity 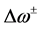 of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP:Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model.
of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP:Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model.
nucleophilic attack on atom k and then that atom acts as an electrophilic 100 species; conversely, when 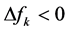 the process is driven by an electrophilic attack over atom k and therefore atom k acts as a nucleophilic species.
the process is driven by an electrophilic attack over atom k and therefore atom k acts as a nucleophilic species.
In 2014, Domingo proposed the Parr functions 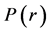 [35] [36] which are given by the following equations:
[35] [36] which are given by the following equations:
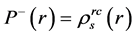 (for electrophilic attacks) and
(for electrophilic attacks) and 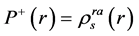 (for nucleophilic attacks) which are related to the atomic spin density (ASD) at the r atom of the radical cation or anion of a given molecule, respectively. The ASD over each atom of the radical cation and radical anion of the molecule gives the local nucleophilic
(for nucleophilic attacks) which are related to the atomic spin density (ASD) at the r atom of the radical cation or anion of a given molecule, respectively. The ASD over each atom of the radical cation and radical anion of the molecule gives the local nucleophilic 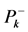 and electrophilic
and electrophilic 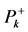 Parr functions of the neutral molecule [37] .
Parr functions of the neutral molecule [37] .
The condensed Fukui functions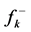 , condensed dual descriptors
, condensed dual descriptors 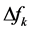 and electrophilic Parr functions
and electrophilic Parr functions 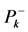 over the N atoms of the amino groups of the Lys residues of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP: Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model are presented in Table 2.
over the N atoms of the amino groups of the Lys residues of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP: Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model are presented in Table 2.
The first thing that can be observed from Table 2 is that the results for the descriptors through calculations based on the Mulliken Population Analysis (MPA) or Hirshfeld Population Analysis (HPA) are roughly the same and that there are not qualitatively differences between them.
However, it can be observed from Table 1 that the chemical reactivity will be
Table 2. The condensed Fukui functions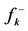 , condensed dual descriptors
, condensed dual descriptors 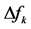 and electrophilic Parr functions
and electrophilic Parr functions 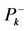 over the N atoms of the amino groups of the Lys residues of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP:Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model. MPA: Mulliken Population Analysis, HPA: Hirshfeld Population Analysis.
over the N atoms of the amino groups of the Lys residues of the optimized conformers of the KD4, KD2, KH4, RKD4 and K14 model peptides calculated with the ONIOM method (MN12SX/Def2TZVP:Amber) using water as solvent simulated with the SMD parametrization of the IEF-PCM model. MPA: Mulliken Population Analysis, HPA: Hirshfeld Population Analysis.
different for the different conformational structures of the model peptides. The amino groups of the Lys residues in the model helical peptides make these systems to behave as electrodonating molecules in the glycation reaction. Thus, KD4-3 and KD4-2 will be more reactive than KD4-1. The same trend is observed for the chemical hardness , the global electrophilicity
, the global electrophilicity 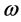 and the net electrophilicity
and the net electrophilicity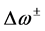 . If we now turn to Table 2, it can be seen that the values of the condensed dual descriptor
. If we now turn to Table 2, it can be seen that the values of the condensed dual descriptor 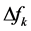 and the electrophilic Parr functions
and the electrophilic Parr functions 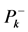 over the N atoms of the amino groups of the Lys residues are larger for the KD4-3 and KD4-2 model peptides than for the KD4-1 system. Therefore, the global and local Conceptual DFT descriptors predict the same behavior and can discriminate between the different conformational structures.
The same conclusions may be extracted from the values of the Conceptual DFT descriptors from Table 1 and Table 2 for the other model helical peptides.
over the N atoms of the amino groups of the Lys residues are larger for the KD4-3 and KD4-2 model peptides than for the KD4-1 system. Therefore, the global and local Conceptual DFT descriptors predict the same behavior and can discriminate between the different conformational structures.
The same conclusions may be extracted from the values of the Conceptual DFT descriptors from Table 1 and Table 2 for the other model helical peptides.
Indeed, the sites for glycation will be also the preferred sites for protonation. Thus, the pKa’s of the Lys residues will be also dependent on the conformational structures of the model peptides. Moreover, the trend in the pKa of the different conformers may be predicted in terms of the local hypersoftness (LHS), which is a local reactivity descriptor that has been defined so that it permits to measure local reactivitiesaccording to the molecular size [30] [32] . The working equation is expressed as follows: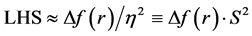 , where S stands for the global softness [4] [38] [39] . As the local hypersoftness can be condensed over the atomic sites, the condensed local hypersoftness is simply computed as
, where S stands for the global softness [4] [38] [39] . As the local hypersoftness can be condensed over the atomic sites, the condensed local hypersoftness is simply computed as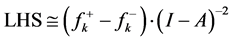 . The procedure is explained as follows:
. The procedure is explained as follows: 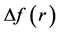 is expressed in atomic units, meanwhile S is measured in mili eV raised to the power of −1, however before performing the multiplication, the mili factor is turned back into 10−3 and then S is raised to the power of 2; the resulting value uses the unit mili eV raised to the power of −2, meaning m(eV−2); the paren- thesis is put in order to make clear that the prefix mili is not raised to the power of -2.
By considering the most reactive conformer of each model peptide, the following trend for the pKa’s of the Lys residues can be found: KD4 ≈ KD2 > KH4 > RKD4 > K14. That is, although the exact value of the pKa cannot be predicted, a qualitative information about their relative ordering can be established by considering the global and local Conceptual DFT descriptors.
is expressed in atomic units, meanwhile S is measured in mili eV raised to the power of −1, however before performing the multiplication, the mili factor is turned back into 10−3 and then S is raised to the power of 2; the resulting value uses the unit mili eV raised to the power of −2, meaning m(eV−2); the paren- thesis is put in order to make clear that the prefix mili is not raised to the power of -2.
By considering the most reactive conformer of each model peptide, the following trend for the pKa’s of the Lys residues can be found: KD4 ≈ KD2 > KH4 > RKD4 > K14. That is, although the exact value of the pKa cannot be predicted, a qualitative information about their relative ordering can be established by considering the global and local Conceptual DFT descriptors.
5. Conclusion
The preferred glycation sites of several model helical peptides have been established by resorting to the calculation of some Conceptual DFT descriptors like the Fukui function indexes, the condensed dual descriptor 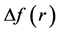 and the electrophilic and nucleophilic Parr functions. The results were obtained within the framework of QM:MM calculations performed through the ONIOM method in the presence of water as a solvent.
The pKa’s of the different Lys residues could be qualitatively predicted on the light of the obtained values for the Conceptual DFT descriptors. Therefore, the techniques presented in this work can be regarded as a practical computational tool for the prediction of the preferred glycation sites of peptides and proteins and for a qualitative description of the pKa’s associated to the ionizable side-chain groups.
and the electrophilic and nucleophilic Parr functions. The results were obtained within the framework of QM:MM calculations performed through the ONIOM method in the presence of water as a solvent.
The pKa’s of the different Lys residues could be qualitatively predicted on the light of the obtained values for the Conceptual DFT descriptors. Therefore, the techniques presented in this work can be regarded as a practical computational tool for the prediction of the preferred glycation sites of peptides and proteins and for a qualitative description of the pKa’s associated to the ionizable side-chain groups.
Acknowledgements
This work has been partially supported by CIMAV, SC and Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) through Grant 219566/2014 for Basic Science Research and Grant 265217/2016 for a Foreign Sabbatical Leave. Daniel Glossman-Mitnik conductedthis work while a Sabbatical Fellow at the University of the Balearic Islands from which support is gratefully acknowledged. This work was cofunded by the Ministerio de Economía y Competitividad (MINECO) and the European Fund for Regional Development (FEDER) 175 (CTQ2014-55835-R).
Cite this paper
Frau, J. and Gloss- man-Mitnik, D. (2017) Comparative Study of the Chemical Reactivity of Helical Peptide Models for Protein Glycation. Computational Chemistry, 5, 65-73. https://doi.org/10.4236/cc.2017.52006
References
- 1. Venkatraman, J., Aggarwal, K. and Balaram, P. (2001) Helical Peptide Models for Protein Glycation: Proximity Effects in Catalysis of the Amadori Rearrangement. Chemistry&Biology, 8, 611–625.
https://doi.org/10.1016/S1074-5521(01)00036-9 - 2. Howard, M.J., Smales, C.M. (2005) NMR Analysis of Synthetic Human Serum Albumin α-Helix 28 Identifies Structural Distortion upon Amadori Modification. The Journal of Biological Chemistry 280 (24), 22582–22589.
https://doi.org/10.1074/jbc.M501480200 - 3. Povey, J., Howard, M.J., Williamson, R.A. and Smales, C.M. (2008) The Effect of Peptide Glycation on Local Secondary Structure.Journal of Structural Biology 161, 151–161.
https://doi.org/10.1016/j.jsb.2007.10.004 - 4. Parr, R. and Yang, W. (1989) Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York.
- 5. Geerlings, P., De Proft, F. andLangenaeker, W. (2003) Conceptual Density Functional Theory. Chemical Reviews103, 1793–1873.
https://doi.org/10.1021/cr990029p - 6. Chattaraj, P:K. (Ed.) (2009) Chemical Reactivity Theory-A Density Functional View, CRC Press. Taylor & Francis Group, Boca Raton.
- 7. Glossman-Mitnik, D. (2013) A Comparison of the Chemical Reactivity of Naringenin Calculated with M06 Family of Density Functionals.Chemistry Central Journal 7, 155–161.
https://doi.org/10.1186/1752-153X-7-155 - 8. Martínez-Araya, J.I., Salgado-Morán,G. and Glossman-Mitnik, D. (2013) Computational Nanochemistry Report on the Oxicams-Conceptual DFT and Chemical Reactivity.Journal of Physical Chemistry B117 (21), 6639–6651.
https://doi.org/10.1021/jp400241q - 9. Glossman-Mitnik, D. (2013) Computational Nanochemistry Study of the Chemical Reactivity Properties of the Rhodamine B Molecule, Procedia Computer Science 18, 816–825.
https://doi.org/10.1016/j.procs.2013.05.246 - 10. Martinez-Araya, J.I., Salgado-Moran, G. and Glossman-Mitnik, D. (2013) Computational Nutraceutics: Chemical Reactivity Properties of the Flavonoid Naringin by Means of Conceptual DFT. Journal of Chemistry 2013 (850297), 8 pages.
- 11. Glossman-Mitnik, D. (2014) Chemical Reactivity Theory within DFT Applied to the Study of the Prunin Flavonoid. European International Journal of Science and Technology, 3, 195-207.
- 12. Glossman-Mitnik, D. (2014) Computational Chemistry of Natural Products: A Comparison of the Chemical Reactivity of Isonaringin Calculated with the M06 Family of Density Functionals. Journal of Molecular Modeling, 20, 1-7.
https://doi.org/10.1007/s00894-014-2316-3 - 13. Frau, J., Munoz, F. and Glossman-Mitnik, D. (2016) A Molecular Electron Density Theory Study of the Chemical Reactivity of Cis- and Trans-Resveratrol. Molecules, 21, 1650.
https://doi.org/10.3390/molecules21121650 - 14. Parr, R. and Yang, W. (1984) Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. Journal of the American Chemical Society, 106, 4049-4050.
https://doi.org/10.1021/ja00326a036 - 15. Parr, R., Szentpaly, L. and Liu, S. (1999) Electrophilicity Index. Journal of the AmericanChemical Society, 121, 1922-1924.
https://doi.org/10.1021/ja983494x - 16. Gazquez, J.L., Cedillo, A. and Vela, A. (2007) Electrodonating and Electroaccepting Powers. Journal of Physical Chemistry A, 111, 1966-1970.
https://doi.org/10.1021/jp065459f - 17. Chattaraj, P.K., Chakraborty, A. and Giri, S. (2009) Net Electrophilicity. Journal of Physical Chemistry A, 113, 10068-10074.
- 18. Avogadro: An Open-Source Molecular Builder and Visualization Tool, Version 1.2.0
- 19. Hanweel, M., Lonie, D., Vandermeersch, T., Zurek, E. and Hutchison, G. (2012) Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. Journal of Cheminformatics, 4, 17.
https://doi.org/10.1186/1758-2946-4-17 - 20. Wang, R.M., Wolf, J.W., Caldwell, P.A. and Kollman, D.A. (2004) Case, Development and Testing of a General AMBER Force fFeld. Journal of Computational Chemistry, 25, 1157-1174.
https://doi.org/10.1002/jcc.20035 - 21. Chung, I.W., Sameera, W.M.C., Ramozzi, R., Page, A.J., Hatanaka, M., Petrova, G.P., Harris, T.V., Li, X., Ke, Z., Liu, F., Li, H.B., Ding, L. and Morokuma, K. (2015) The ONIOM Method and Its Applications. Chemical Reviews, 115, 5678-5796.
https://doi.org/10.1021/cr5004419 - 22. Marenich, A., Cramer, C. and Truhlar,D. (2009) Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. Journal of Physical Chemistry B, 113, 6378-6396.
https://doi.org/10.1021/jp810292n - 23. Peverati, R. and Truhlar, D.G. (2012) Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Physical Chemistry Chemical Physics, 14, 16187-16191.
https://doi.org/10.1039/c2cp42576a - 24. Weigend, F. and Ahlrichs, R. (2005) Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Physical Chemistry Chemical Physics, 7, 3297-3305.
https://doi.org/10.1039/b508541a - 25. Weigend, F. (2006) Accurate Coulomb-fitting Basis Sets for H to R. Physical Chemistry Chemical Physics, 8, 1057-1065.
https://doi.org/10.1039/b515623h - 26. Cornell, W., Cieplak, B., Bayly, C., Gould, I., Merz, K., Ferguson, D., Spellmeyer, D, Fox, T., Caldwell, J. and Kollman, P. (1995) A Second Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic Molecules. Journal of the American Chemical Society, 117, 5179-5197.
https://doi.org/10.1021/ja00124a002 - 27. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., and Fox, D.J. (2009) Gaussian 09, Revision E.01. Gaussian Inc., Wallingford.
- 28. Morell, C., Grand, A. and Toro-Labbé, A. (2005) New Dual Descriptor for Chemical Reactivity. Journal of Physical Chemistry A, 109, 205-212.
https://doi.org/10.1021/jp046577a - 29. Morell, C., Grand, A. and Toro-Labbé, A. (2006) Theoretical Support for Using the f(r) Descriptor. Chemical Physics Letters, 425, 342-346.
- 30. Cárdenas, C., Rabi, N., Ayers, P., Morell, C., Jaramillo, P. and Fuentealba, P. (2009) Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hyper Softness, and Electrostatic Potential. Journal of Physical Chemistry A, 113, 8660-8667.
https://doi.org/10.1021/jp902792n - 31. Toro-Labbé, A. (2007) Theoretical Aspects of Chemical Reactivity. Vol. 19, Elsevier Science, Amsterdam.
- 32. Ayers, P., Morell, C., De Proft, F. and Geerlings, P. (2007) Understanding the Woodward-Hoffmann Rules by Using Changes in Electron Density, Chemistry—A European Journal, 13, 8240-8247.
https://doi.org/10.1002/chem.200700365 - 33. Morell, C., Ayers, P., Grand, A., Gutiérrez-Oliva, S. and Toro-Labbé, A. (2008) Rationalization of the Diels-Alder Reactions through the Use of the Dual Reactivity Descriptor f(r). Physical Chemistry Chemical Physics, 10, 7239-7246.
https://doi.org/10.1039/b810343g - 34. Morell, C., Hocquet, A., Grand and Jamart-Gregoire, B. (2008) A Conceptual DFT Study of Hydrazino Peptides: Assessment of the Nucleophilicity of the Nitrogen Atoms by Means of the Dual Descriptor f(r). Journal of Molecular Structure: THEOCHEM, 849, 46-51.
- 35. Domingo, L.R., Pérez, P. and Sáez, J. (2013) Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Advances, 3, 1486-1494.
https://doi.org/10.1039/C2RA22886F - 36. Chamorro, E., Pérez, P. and Domingo, L.R. (2013) On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chemical Physics Letters, 582, 141-143.
- 37. Domingo, L.R., Ríos-Gutiérrez, M. and Pérez, P. (2016) Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules, 21, 748.
https://doi.org/10.3390/molecules21060748 - 38. Pearson, R. (1993) The Principle of Maximum Hardness. Accounts of Chemical Research, 26, 250-255.
https://doi.org/10.1021/ar00029a004 - 39. Chermette, H. (1999) Chemical Reactivity Indexes in Density Functional Theory. Journal of Computational Chemistry, 20, 129-154.
https://doi.org/10.1002/(SICI)1096-987X(19990115)20:1<129::AID-JCC13>3.0.CO;2-A