Computational Chemistry
Vol.2 No.1(2014), Article ID:41817,5 pages DOI:10.4236/cc.2014.21001
Structure of Flavones and Flavonols. Part II: Role of Position on the O-H Bond Dissociation Enthalpy
Department of Chemistry, South-West University “NeofitRilski”, Blagoevgrad, Bulgaria.
Email: *jivko_av@swu.bg
Copyright © 2014 Maria Hr. Vakarelska-Popovska, Zhivko As. Velkov. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Maria Hr. Vakarelska-Popovska, Zhivko As. Velkov. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received September 9th, 2013; revised October 23rd, 2013; accepted November 6th, 2013
KEYWORDS
DFT; BDE; EHOMO; Maximum Spin Density; Flavones; Flavonols
ABSTRACT
The enthalpies of bond dissociations of the hydroxyl groups, the energy of the highest occupied molecular orbitals and the corresponding radicals spin densities of all possible ten isomeric mono-hydroxyl flavones are calculated by means of 3-parameter Becke, Lee, Yang and Parr functional. The structural factors affecting these descriptors of radical-scavenging activity are outlined. The results obtained show that the hydroxyl groups in ring A and C are more involved in the process of a radical-capturing than recently believed.
1. Introduction
Flavonoids are among the most widely studied groups of organic compounds [1]. The reason for the great interest is their antioxidant activity [2] and the various biological actions related with that [3]. Flavonoids have continuously been studied in the recent years for therapeutic activity against the most widespread diseases in the world—cardiovascular diseases, cancer, etc. [4].
Many structural studies of various compounds from this group have been published lately [5-7]. Increasingly sophisticated quantum-chemical methods and high orbital bases are used in them. The aim of these structural studies is to find appropriate correlations between structure and activity that explain the biochemical mechanisms of action of the test substances and rationalize the synthesis of new pharmacologically interesting compounds [8-10].
Since the biological activity of flavonoids is associated with the antioxidant activity, the structural studies are primarily focused on the ability of flavonoids to react with active radicals according to the reaction:
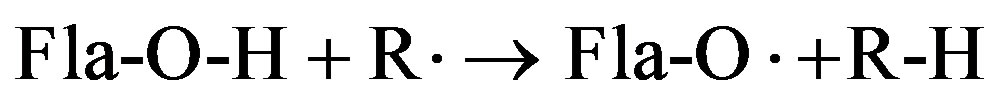
As showed in Figure 1, the flavones are a type of flavonoids with conjugated p-electron system spread accross the whole molecule, which is an important structural feature for the radical-scavenging activity. So far, only natural flavones which usually possess two or more functional groups have been investigated. In these cases, it is difficult to determine what the impact of the various structural factors on the O-H bond dissociation enthalpy (BDE) and the ability of molecules to react with radicals is.
In this study, we will stress on the effect of the position of the hydroxyl group in mono-hydroxy flavones on its ability to react with radicals. For this purpose the descriptors associated with the ability of flavones to donate a hydrogen atom are calculated—BDE [11], the energy of the highest occupied molecular orbital (EHOMO) [12], as well as the stability of the radicals obtained in the course of reaction—maximum spin density (MSD). The last descriptor (MSD) assesses the conversion degree [13] of the reaction between a radical-scavenger and an active radical. BDE describes the ability of a radical-scavenger to enter in a bimolecular homolytic reaction, while EHOMO predicts the rate of reaction determined by electron donation ability.

Figure 1. Basic structure of flavones [2-(2'-phenyl)-chromen- 4-one].
All ten isomeric mono-hydroxyl flavones will be investigated.
2. Computational Details
The calculations were carried out using the DFT, as implemented in the Gaussian09 program package [14]. The optimization of the geometry was performed with the Becke 3-parameter hybrid exchange functional combined with the Lee-Yang-Parr correlation functional (B3LYP) with the standard 6-31G(d,p) basis set [15]. The optimization was achieved without any geometry constraints. For all structures the harmonic vibrational frequencies were computed to confirm the true minima on the calculated potential surface.
The use of the 6-31G(d,p) orbital basis is a reasonable compromise between accuracy and computational cost for the set of investigated molecules and the corresponding radicals. This basis has been previously used for calculations of radical-scavenging activity descriptors [16]. Some of results are confirmed experimentally [17].
All possible intra-molecular interactions were taken into account in the initial geometries.
Solvent effects on the calculated structures were investigated with the self-consistent reaction filed (SCRF) method, via the polarized continuum method (PCM) [18]. Full geometry optimization in water (ε = 78.3553) was performed at the same level of theory as in the gas phase.
BDE was calculated as a difference in total enthalpy of the corresponding radical (formed after H abstraction), hydrogen atom and the flavone, according to:
 where H denotes the enthalpies of the investigated radicals, compounds and hydrogen.
where H denotes the enthalpies of the investigated radicals, compounds and hydrogen.
3. Results and Discussion
In the first part of our investigation [19] we found that BDE of the O-H groups in mono-hydroxyl flavones in vacuum changes in the following order: 2' < 8 < 6 < 4' < 3 < 5' < 3' < 7 < 6' < 5.
As mentioned above, the BDE is calculated as a difference in the enthalpy of the radical and the hydrogen atom on the one hand and the corresponding flavone on the other. Consequently, all structural features which stabilize the flavones and destabilize the corresponding radicals give rise to an increase of BDE and vice versa.
Therefore, the internal molecular factors for that order of the O-H BDE series are two:
• Transfer of electron density from the hydroxyl groups to the carbonyl oxygen depends on the position of the hydroxyl group. This determines the differences in the stability of various flavones. It is easy to realize that the mono-hydroxyl flavones with hydroxyl group at 5, 7, 2', 4' and 6' position have better delocalization and are more stable. Hence the O-H BDE must be greater therein.
• The second reason for the enhancement of O-H BDE is the intra-molecular hydrogen bond with the hydroxyl group hydrogen. It stabilizes additionally the flavones but is missing in the radicals. An intra-molecular electrostatic interaction engaging the O-H group oxygen causes the decrease of BDE.
Let us now see how the strength of the O-H bonds depends on the intra-molecular factors (Table 1).
Although the hydroxyl group at position 3 is not of phenolic type, it has the lowest BDE in solvent. In vacuum the BDE of this group is by 2 kcal/mol higher than that of the hydroxyl group with the lowest BDE (2'-isomer). The reason for the low BDE is in the electrostatic interaction between the oxygen of the hydroxyl group and the hydrogen at the 2' position which stabilizes both the flavone and the radical. The polarizing effect of water stabilizes 3-hydroxy flavone by 5.020 kcal/mol and the corresponding radical by 8.158 kcal/mol. As a result BDE decreases by 3.211 kcal/mol. The radical is strictly flat and more polarizable than the molecule.
The next lowest is the BDE of the hydroxyl group at site 8 in ring A. This position does not allow an effective hydroxyl-to-carbonyl electron density delocalization. Moreover, the hydrogen of the hydroxyl group does not form a strong stabilizing hydrogen bond with the oxygen at position 1. Here, the decrease in the BDE in PCM is only 0.503 kcal/mol. It is noteworthy that the radicals obtained after hydrogen atom abstraction from 8-hydroxy flavone is flat unlike the molecule.
At 1.394 kcal/mol from the lowest BDE is that of the hydroxyl group located at position 2'. The water media decreases the BDE by 0.274 kcal/mol. The place of this hydroxyl group in the BDE row in vacuum and PCM is determined by the stabilization of the radicals through electrostatic interaction between the oxygen of the hydroxyl group and the hydrogen in position 3.
The 4th place in the BDE row belongs to the hydroxyl group located at position 6 in ring A, which is on the third place in vacuum. Water environment lowers the

Table 1. Enthalpies of mono-hydroxyl flavones and O-H BDE.
BDE value by 0.587 kcal/mol. The place in the first part of the row is due to the high enthalpy of the flavone. In vacuum, this isomer is one of the three flavones with highest enthalpies.
At the 5th place in the BDE row is the hydroxyl group located at the position 4'. This group, in conjunction with a hydroxyl group at position 3' is connected with antioxidant activity of flavonoids [5,7,17,20]. Stabilization in PCM is negligible. This group is not so prone to homolytic dissociation. This means that the presence of two adjacent hydroxyl groups and easy formation of ortho-quinones is the real cause of the high antioxidant activity, i.e. or the reaction proceeds via electron transfer, as we will see below.
The BDEs of the remaining three hydroxyl groups in ring B: 6', 3' and 5' come next in this order in solvent (in vacuum the order is different). In fact only the position 6' of the following three mono-hydroxy flavones may provide stability at the expense of efficient conjugation of the hydroxyl group to the carbonyl group. Тhe hydrogen bond forming 6-membered ring with the oxygen at position 1 increases the BDE.
The next value of BDE belongs to the hydroxyl group located at position 7. This proves clearly that the withdrawal of the electronic density from the hydroxyl group to the carbonyl oxygen increases the BDE of the hydroxyl group. The increase in BDE in the presence of water/polar environment is 1.358 kcal/mol because the stabilization of the flavone is better than in the radical.
The hydroxyl group located at position 5 has the largest BDE. The two important factors stabilizing monohydroxyl flavones operate here: stabilization through delocalization and stabilization by intra-molecular hydrogen bonding. Obviously, the second factor is more important because the BDE of this hydroxyl group is significantly larger than the previous one and all the rest. This hydrogen bond is very strong. The distance between the hydroxyl hydrogen and the carbonyl oxygen is only 1.682 Å.
Significant reduction of BDE in polar medium was found in two isomers: 3- and 5-hydroxyflavone. These are the mono-hydroxyl flavones in which the hydroxyl groups participate in intra-molecular hydrogen bonds forming 5-membered rings. In the other compounds the change of BDEs is either negligible or negative due to better stabilization of the radicals than the hydroxyflavone.
In some mono-hydroxyl flavones BDEs increase in water. The increase is most prominent in 7-hydroxy flavone.
Table 2 comprises two other descriptors of the investigated compounds that may be associated with the radical-scavenging activity of the tested compounds.
Between MSD and BDE, however there is a significant linear correlation (BDE = 0.2865MSD + 0.0173) within the confidence interval of 99% for the results taking into account the polarising effect of the water (R = 0.818 at Rcritical value = 0.765 for 8 degree of freedom) [21]. The increase of MSD increases BDE. It is a quite natural conclusion confirming the above rule: isomers with more stable radicals have lower BDE.
EHOMO shows the tendency of the compound to donate electrons. It appears that the reactivity decreases in the following order:
1) in vacuum:
3-ОН < 5-ОН < 6-ОН < 4'-ОН < 2'-ОН < 8-ОН < 5'-ОН < 6'-ОН < 7-ОН < 3'-ОН
2)in water:
3-ОН < 4'-ОН < 6-ОН < 5-ОН < 2'-ОН < 8-ОН < 6'-ОН < 5'-ОН < 3'-ОН < 7-ОН.
Comparison between EHOMO and BDE is not entirely correct since the first one is a microscopic characteristic
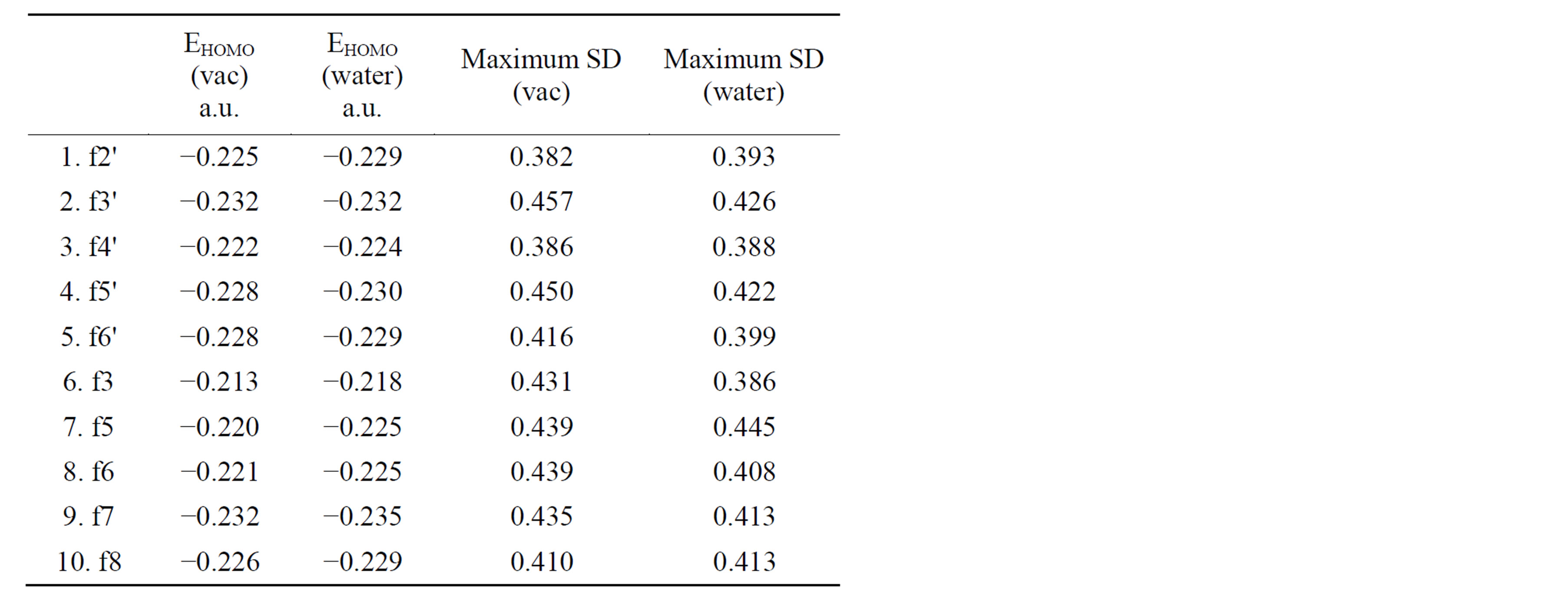
Table 2. Energies of HOMOs and maximum spin densities.
and the second is a macroscopic. The main contribution to the enthalpy of the states, however, comes from the electronic energy and that allows us to make a qualitative argument. A comparison of these two descriptors for each compound indicates that the energy of electron separation is significantly higher than the energy for the separation of hydrogen even in water medium. This means that the separation of an electron from hydroxy flavone requires a preliminary deprotonation of the hydroxyl group. It is always easier to take an electron from conjugate bases than from the corresponding conjugate acids. In this study, however, we have not included the corresponding anions.
The row of reduction of reactivity of the mono-hydroxy flavones optimized in vacuum and ordered by EHOMO differs significantly from the above BDE row. Therefore, the intra-molecular structural factors which regulate the ability of mono-hydroxy flavones to donate H-atom do not determine the ability of the compounds to lose electrons.
The row of increasing of EHOMO in water is more interesting because the reactions passing through an electron transfer can be realized mainly in polar medium. On the other hand, the deprotonation of phenolic hydroxyl groups is possible only in water.
3-hydroxy flavone is again the most active in the water environment followed by 4'-isomer. This result coincides completely with the view in the literature that biologically active flavonoids possess hydroxyl groups of 3 and 4'- positions [5,20,22,23]. Consequently that is the case when assessing radical-scavenging activity passing through an electron transfer followed by a proton transfer mechanism. The findings of the authors show that this is the mechanism in which flavonoids react with active radicals.
4. Conclusions
Factors of BDE increase in mono-hydroxy are: 1) stabilization of hydroxy flavones through electron density transfer from the hydroxyl group towards the carbonyl group at position 4; or 2) through intra-molecular hydrogen bonds with the hydrogen from the hydroxyl group; 3) BDE decreases in the isomers having electrostatic interaction between the oxygen from the hydroxyl group and a hydrogen from the adjacent ring, because the resulting radical is stabilized.
The results obtained for EHOMO show that flavones could participate in reactions whose rate is controlled by the separation of an electron only after deprotonation of the phenolic hydroxyl groups.
REFERENCES
- Ell. Middleton Jr., Ch. Kandaswami and Th. C. Theoharides, “The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer,” Pharmacological Reviews, Vol. 52, No. 4, 2000, pp. 673-751.
- P.-G. Pietta, “Flavonoids as Antioxidants,” Journal of Natural Products, Vol. 63, No. 7, 2000, pp. 1035-1042. http://dx.doi.org/10.1021/np9904509
- A. R. Tapas, D. M. Sakarkar and R. B. Kakde, “Flavonoids as Nutraceuticals: A Review,” Tropical Journal of Pharmaceutical Research, Vol. 7, No. 3, 2008, pp. 1089- 1099. http://dx.doi.org/10.4314/tjpr.v7i3.14693
- P. C. H. Hollman and M. B. Katan, “Dietary Flavonoids: Intake, Health Effects and Bioavailability,” Food and Chemical Toxicology, Vol. 37, No. 9-10, 1999, pp. 937- 942. http://dx.doi.org/10.1016/S0278-6915(99)00079-4
- B. F. Rasulev, N. Abdullaev, V. N. Syrov and J. Leszczynski, “A Quantitative Structure-Activity Relationship (QSAR) Study of the Antioxidant Activity of Flavonoids,” QSAR & Combinatorial Science, Vol. 24, No. 5, 2005, pp. 1056-1065. http://dx.doi.org/10.1002/qsar.200430013
- T. Z. Todorova, M. G. Traykov, A. V. Tadjer and Zh. A. Velkov, “Structure of Flavones and Flavonols. Part I: Role of Substituents on the Planarity of the System,” Computational and Theoretical Chemistry, Vol. 1017, 2013, pp. 85-90. http://dx.doi.org/10.1016/j.comptc.2013.05.005
- D. Amic, D. Davidovic-Amic, D. Beslo and N. Trinjastic, “Structure-Radical Scavenging Activity Relationships of Flavonoids,” Croatica Chemica Acta, Vol. 76, No. 1, 2003, pp. 55-61.
- A. D. Yılmaz, T. Coban and S. Suzen, “Synthesis and Antioxidant Activity Evaluations of Melatonin-Based Analogue Indole-Hydrazide/Hydrazone Derivatives,” Journal of Enzyme Inhibition and Medicinal Chemistry, Vol. 27, No. 3, 2012, pp. 428-436. http://dx.doi.org/10.3109/14756366.2011.594048
- H. Shirinzadeh, B. Eren, H. Gurer-Orhan, S. Suzen and S. Ozden, “Novel Indole-Based Analogs of Melatonin: Synthesis and in Vitro Antioxidant Activity Studies,” Molecules, Vol. 15, No. 4, 2010, pp. 2187-2202.
- S. Suzen, S. S. Cihaner and T. Coban, “Synthesis and Comparison of Antioxidant Properties of Indole-Based Melatonin Analogue Indole Amino Acid Derivatives,” Chemical Biology & Drug Design, Vol. 79, No. 1, 2012, pp. 76-83. http://dx.doi.org/10.1111/j.1747-0285.2011.01216.x
- K. R. Korzekwa, J. P. Jones and J. R. Gillette, “Theoretical Studies on Cytochrome P-450 Mediated Hydroxylation: A Predictive Model for Hydrogen Atom Abstractions,” Journal of the American Chemical Society, Vol. 112, No. 19, 1990, pp. 7042-7046. http://dx.doi.org/10.1021/ja00175a040
- T. Koopmans, “Über die Zuordnung von Wellenfunktionen und Eigenwertenzu den Einzelnen Elektroneneines Atoms,” Physica, Vol. 1, No. 1-6, 1934, pp. 104-113.
- Zh. Cheng, J. Ren, Y. Li, W. Chang and Zh. Chen, “Establishment of a Quantitative Structure—Activity Relationship Model for Evaluating and Predicting the Protective Potentials of Phenolic Antioxidants on Lipid Peroxidation,” Journal of Pharmaceutical Sciences, Vol. 92, No. 3, 2003, pp. 475-484. http://dx.doi.org/10.1002/jps.10301
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K.Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. M. Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Lyengar, J. Tomasi, M. Coss, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B.Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc.,Wallingford CT, 2009.
- R. G. Parr and W. Yang, “Density-Functional Theory of Atoms and Molecules,” Oxford University Press, Oxford, 1989.
- K. M. Nikolic, “Theoretical Study of Phenolic Antioxidants Properties in Reaction with Oxygen-Centered Radicals,” Journal of Molecular Structure: THEOCHEM, Vol. 774, No. 1-3, 2006, pp. 95-105. http://dx.doi.org/10.1016/j.theochem.2006.07.017
- S. Riahi, P. Norouzi, Ab. B. Moghaddam, M. R. Ganjali, Gh. R. Karimipour and H. Sharghi, “Theoretical and Experimental Report on the Determination of Oxidation Potentials of Hydroxyanthracene and Thioxanthene Derivatives,” Chemical Physics, Vol. 337, No. 1-3, 2007, pp. 33-38. http://dx.doi.org/10.1016/j.chemphys.2007.06.018
- J. B. Foresman, T. A. Keith, K. B. Wiberg, J. Snoonian and M. J. Frisch, “Solvent Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on Ab Initio Reaction Field Calculations,” Journal of Physical Chemistry, Vol. 100, No. 40, 1996, pp. 16098- 16104. http://dx.doi.org/10.1021/jp960488j
- M. Vakarelska-Popovska and Zh. Velkov, “QuantumChemical Calculation of O-H Bond Dissociation Enthalpy in Flavones,” Proceedings of the 5th International Scientific Conference, Vol. 4, 2013, pp. 10-16.
- S. A. B. E. van Acker, M. J. de Groot, D. J. van den Berg, M. N. J. L. Tromp, G. D. O. den Kelder and W. J. F. van der Vrjgh and A. Bast, “A Quantum Chemical Explanation of the Antioxidant Activity of Flavonoids,” Chemical Research in Toxicology, Vol. 9, No. 8, 1996, pp. 1305- 1312. http://dx.doi.org/10.1021/tx9600964
- M. F. Triola, “Elementary Statistics,” Addison-Wesley, Boston, 2003.
- P. F. Kador and N. E. Sharpless, “Structure-Activity Studies of Aldose Reductase Inhibitors Containing the 4- Oxo-4H-Chromen Ring System,” Biophysical Chemistry, Vol. 8, No. 1, 1978, pp. 81-85. http://dx.doi.org/10.1016/0301-4622(78)85025-X
- P. F. Kador and N. E. Sharpless, “Pharmacophor Requirements of the Aldose Reductase Inhibitor Site,” Molecular Pharmacology, Vol. 24, No. 3, 1983, pp. 521-531.
NOTES
*Corresponding author.