Open Journal of Immunology
Vol.2 No.2(2012), Article ID:20125,6 pages DOI:10.4236/oji.2012.22007
The law of unintended consequences and antibiotics
![]()
1Department of Microbiology and Immunology, Drexel University College of Medicine, Philadelphia, USA;
*Corresponding Author: carol.artlett@drexelmed.edu
2TherimuneX Pharmaceuticals Inc., Doylestown, USA
Received 17 February 2012; revised 8 March 2012; accepted 23 March 2012
Keywords: Antibiotic Resistance; Inflammasome; Microbiota; Emerging Infectious Diseases; Chronic Diseases
ABSTRACT
Antibiotic therapy has been of unquestionable value in terms of saving lives and reducing suffering caused by infectious diseases. However, the widespread and indiscriminant use of antibiotics has not been without significant consequences. Some of these consequences include increased antibiotic resistance in pathogenic strains of bacteria. In addition, the indiscriminate use of antibiotics has recently been shown to alter the host microbiota and this has been associated with chronic disease. However, abandoning the use of antibiotics would be unthinkable. We have been exploring a strategy of modulating the host tolerance/resistance mechanisms as a means to ablate infection. We postulate that the obvious advantage of this therapeutic strategy is that, by targeting host tolerance/resistance mechanisms, the selective pressure for the expression of pathogen resistance is greatly reduced or altogether absent.
1. INTRODUCTION
Alexander Fleming’s discovery of Penicillin in 1928 and the dawn of the “Golden Age of Antibiotics” helped to spawn the pharmaceutical industry as we know it today. Antibiotic therapy has been the mainstay medical modality for treating infections of all bacterial types and has been of unquestionable value in terms of lives saved and suffering alleviated from infectious diseases. The impact of antibiotics on medical practices is not just limited to controlling infectious diseases, as complex surgical procedures such as organ transplantation or joint replacement would be inconceivable without prophylactic antibiotic therapy. However, the widespread and often indiscriminant use of antibiotics in modern industrialized societies has not been without consequences and herein we broadly refer to these consequences as “antibiotic induced pathogenesis”, i.e. the generation of diseases resulting from the excessive use of antibiotics.
2. ANTIBIOTIC-INDUCED PATHOGENESIS
The types of antibiotic induced pathogenesis can be broadly characterized as either arising from the effect of an antibiotic acting directly on the pathogen (pathogencentric) or the effect of an antibiotic on the host’s microbiota (host-centric). As an example of a host-centric antibiotic induced pathogenesis, Clostridium difficile emerged in the 1970’s as an opportunistic pathogen that colonized the colon of patients whose intestinal microflora was decimated by oral Clindamycin therapy [1]. Today antibiotic resistant C. difficile is a major concern in hospital settings with periodic outbreaks occurring in North America and Europe [1].
As an example of the pathogen-centric antibiotic induced pathogenesis, β-lactamase producing Staphylococcus aureus has emerged as a penicillin-resistant strain occurring shortly after the widespread use of penicillin in the 1940’s and the broad use of the β-lactam class of antibiotics selected for β-lactamase producing S. aureus. Today, methicillin-resistant S. aureus (MRSA) is one of the major pathogens in hospital-acquired infections as evidenced from The Center for Disease Control report that more people die yearly from hospital-acquired MRSA infections than HIV in the United States and that hospital-acquired infections in the United States has added an estimated $30 billion in health-care costs annually [2]. MRSA was considered as a problem that was largely confined to hospitals and extended care facilities. However, the possibility of horizontal transfer of resistance from the hospital environment to the community has been a major concern and recently there have been reports of the emergence of a community acquired MRSA (CA-MRSA) that has validated this concern [3-5].
The pathogen-centric type may be the more common and obvious method of antibiotic-induced pathogenesis. This is largely because bacteria have harbored a wide array of antibiotic resistant genes as a natural survival mechanism [6]. In support of this hypothesis, resistance genes for all known antibiotics have recently been reported in the bacterial genome of ancient bacteria [7]. The emergence of antibiotic resistant opportunistic pathogens from environmental reservoirs (Acinetobacter sp. and Pseudomonas sp.) serves to make the point that the environment is a large reservoir of antibiotic resistance genes that can be horizontally transferred to susceptible pathogens. For example, it is now believed that the multi-drug resistant strains of bacterial pathogens, Mycobacterium tuberculosis and Escherichia coli being just two examples, have acquired resistance factors via horizontal transfer of resistance genes from other bacteria species (reviewed in [8]). The recent report of an isolated strain of Neisseria gonorrhea that is resistant to all known antibiotics may be another example of acquired resistance from inter-species gene transfer [9]. In fact it now appears that the genome of all bacteria species is a huge gene-pool upon which all bacteria species can draw. Thus, it is readily apparent that the massive selection pressure provided by widespread and indiscriminate use of antibiotics has selected for the resistance factors in modern pathogenic bacteria and that nature is taking care of the rest through vertical and horizontal transfer of resistance genes within and across bacterial species.
3. ANTIBIOTIC-INDUCED CHRONIC DISEASE
If that is not sobering enough, host-centric antibioticinduced pathogenesis is far more insidious with a potential for long-term consequences. Humans are not gnotobiotic, except perhaps at birth, but are hosts to a diverse population of beneficial microorganisms—the human microbiota. The microbiota in an individual human evolves in a more or less orderly manner commencing with the natural birth process and continuing throughout life that is influenced by the individual’s genetics, diet, and environmental exposure factors. Of the more than 50 bacterial phyla only 4 are found in the human microbiome (Actinobacteria, Bacteriodetes, Firmucutes, and Protobacteria) and are located predominantly in the mucosal and dermal compartments [10]. This suggests that coevolution and co-adaptation over hundreds of thousands of years between humans and their microbiome have limited the bacterial diversity to the current steady-state.
The steady-state dynamics of the human microbiome cannot be fully appreciated when thought of as a static system, nor as a simple equilibrium. The human microbiota facilitates a wide variety of physiological processes (vitamin acquisition and nutrient absorption, tissue integrity, neuronal function, and immune regulation) and the microbiota occupies multiple anatomical compartments. Blaser and Kirschner introduced the concept of nested equilibria and invoked the Nash equilibrium to describe physiological interaction with the microbiota [11]. Adopted from game theory and applied to complex biological interactions, the Nash equilibrium represents a set of boundary conditions that if transgressed result in a severe penalty that nullifies any competitive advantage arising from the transgression. Blaser and Kirschner conceptualized a series of nested Nash equilibria in which the boundary conditions of one compartment contribute to the boundary conditions of the others. Thus, the nested ecosystems provide stability and adaptive flexibility (tolerance) between the host and the microbiota to tolerate potential pathogens; E. coli in the intestinal tract or S. aureus on the dermis and nasal mucosa for example. Viewed through a lens of nested Nash equilibria we can begin to appreciate the potential consequences of hostcentric antibiotic induced pathogenesis.
As noted earlier, alteration in the intestinal microbiota by Clindamycin therapy enabled antibiotic resistant C. difficile to colonize the vacated niche. An example of more insidious consequences may be the steady disappearance of Helicobacter pylori from the human gastric microbiota. Blaser and Falko postulate that the concomitant rise in esophageal reflux, Barrett’s esophagus, and esophageal adenocarcinoma may be directly related [12].
Th17 cells are a CD4+ T-cell phenotype and these cells are a critical subset of effector cells that regulate the immune response and confer protection against a wide variety of bacterial and fungal pathogens (reviewed in [13]). High numbers of Th17 effector cells have been strongly associated with several chronic inflammatory diseases such as Crohn’s disease, colitis, rheumatoid arthritis, asthma, and multiple sclerosis [14]. In addition, the regulatory T-cell phenotype (Tregs) is a differentiated lineage of CD4+ T-cells that play a central role in immune tolerance. These cells are delineated by the Foxp3+ phenotype. Foxp3+ Tregs have been shown to be substantially reduced in the stomach of patients devoid of H. pylori and epidemiological studies established that H. pylori positive individuals have much lower incidences of asthma and other related allergic conditions (reviewed in [15]).
Furthermore, there is an inverse relationship between Tregs and Th17 cells and the plasticity of the Treg-Th17 balance in humans has been recently described [16]. We speculate that the shifting of the Treg-Th17 balance towards the Th17 phenotype in the gastric compartment could induce a global Th17 response that would provide a mechanistic explanation for the higher incidence of asthma in individuals devoid of a gastric H. pylori population. Experimental and clinical validation of this hypothesis would validate the nested Nash equilibrium model and demonstrate that perturbation of the microbiota (i.e. eradication of H. pylori) in the gastric mucosal compartment effects the boundary conditions and the Treg-Th17 balance in another (the respiratory tract) leading to disease (asthma).
4. ANTIBIOTIC-INDUCED ALTERATION OF THE GUT MICROBIOTA AND OBESITY
Another example of a host-centric antibiotic induced pathogenesis may be the epidemic rise in obesity—particularly in children. The intestinal microbiota in obese patients has been found to be constitutively different from that found in normal weight individuals. Morbidly obese individuals have an increased proportion of H2- producing Prevotellaceae and H2-utilizing methanogenic Archaea. Similar results have been observed in obese mice as compared to their normal weight counterparts [17]. These observations have prompted the hypothesis that alteration of the intestinal microbiota in favor of H2- producing and H2-utilizing organisms enables a much more efficient caloric uptake in obese individuals.
Sub-therapeutic doses of broad-spectrum antibiotics have been used in feed additives in food animal agriculture for decades because they induce increased weight gain in animals, particularly in the young [18]. Thus, for several generations, the population in industrialized societies has been ingesting sub-therapeutic doses of broadspectrum antibiotic residues from a diet of dairy, meat, and poultry products. Could chronic exposure to subtherapeutic doses of broad-spectrum antibiotic residues in the diet be having a similar effect on children as it does on food animals? Considering the long-term health consequences of obesity to the individual and the increasing burden on the health-care system caused by obesity related health issues, this is a question that urgently needs to be answered. If credible experimental research and epidemiological studies support this hypothesis, then resolving the obesity epidemic by banning use of antibiotics as a feed-additive in agriculture is a simple solution to this major public health problem.
5. VACCINE-INDUCED PATHOGENESIS
The use of pathogen-specific vaccines can have consequences that are similar to antibiotic induced pathogenesis. Streptococcus pneumoniae is tolerated as a normal constituent in the nasal microbiota without causing disease. A more virulent, encapsulated strain, however, causes upper respiratory tract infections, pneumonia, and occasionally fatal endocarditis or meningitis. A polyvalent vaccine against the most virulent subtypes of pneumococcus is effective in reducing the incidence of serious infection in at-risk populations such as college students and military recruits [19]. Because, S. aureus and S. pneumoniae are ecological competitors [20], the vacating S. pneumoniae is being replaced by S. aureus in the community [21]. The net effect of a vaccination against S. pneumoniae is the replacement of one tolerated bacteria with a pathogen and the epidemic rise in the incidence of CA-MRSA infections.
6. ALTERNATIVE APPROACHES
Abandoning the use of antibiotics is an unthinkable answer. However, the development of new antibiotics has slowed considerably in recent years, in a large part, due to the long lead-time from discovery-to-market for new molecular entities, the high failure rates of new drug candidates, the short treatment period, and the low reimbursement rates for antibiotics. Thus, medical practitioners are left in an ever-widening chasm between the emergence of new antibiotic resistant pathogens and a dearth of therapeutic alternatives. New approaches such as the development of inhibitors of bacterial resistance factors for use in combination therapy with existing antibiotics have promise as a near-term solution [22], but new chemical entities exploiting these strategies have yet to enter clinical trials.
We have been exploring a strategy of modulating the host tolerance/resistance mechanisms as a means to ablate infection from progressing to disease. Mammals have evolved tolerance mechanisms to accommodate beneficial microorganisms as well as potential pathogens, and they also have evolved resistance mechanisms to reduce the pathogen burden during infection. These two types of mechanisms have been reviewed by Schneider and Ayers [23] and they classify the regulators of tolerance and resistance into three basic classes. The first class, comprises resistance and tolerance that are linked and their effects are opposite. Reactive oxygen species is one example of this class. The second class is comprised of regulators that control both resistance and tolerance in an either/or manner and are therefore separable as opposed to absolute linkage in the class one effector molecules. The third class is comprised of regulators that effect only tolerance. The inflammasome signaling platforms represent class two and class three regulators.
The inflammasome comprises a family of cytosolic receptors called NOD-like receptors (NLR) that are involved in innate immune recognition of pathogen associated molecular patterns as well as intracellular and extracellular damage associated molecular patterns (reviewed in [24]). Thus far, more than 20 inflammasomes have been identified and many are present in nearly every cell type. NLRP3 is the most extensively studied inflammasome and has been found to be activated by a diverse range of stimuli including microbial derived pro-
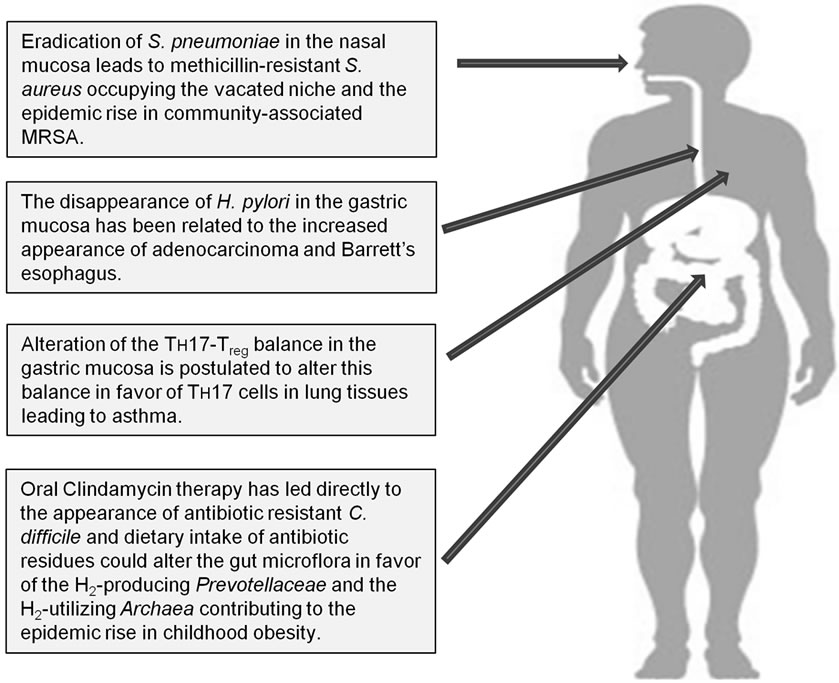
Figure 1. Potential long-term consequences of broad-spectrum antibiotic usage.
ducts [25-28], environmental factors [29-31] and endogenous molecules [32-35]. The NLRP3 inflammasome would represent a class two regulator of resistance/tolerance. The NLRP6 inflammasome, on the other hand, has been recently shown to regulate the composition of the microbiota in the colon and therefore would represent a class three regulator of tolerance [36].
We have described the regulation of the NLRP3 inflammasome as an effective means to prevent Grampositive and Gram-negative bacterial infections from progressing to disease, enhancing tolerance; and abrogating disease progression in an experimentally established MRSA infection or a clinically established multifactorial bacterial infection leading to enhanced resistance [37]. The obvious advantage of this therapeutic strategy is that, by targeting the host tolerance/resistance mechanisms, the selective pressure for the expression of pathogen resistance is greatly reduced or altogether absent. This is not to say that such drugs will replace antibiotics. Rather, we envision a medical modality that will utilize these anti-infective drugs to prophylactically reduce the incidence of infections that progress to disease. Adjunctive therapy with the next generation of highly selective, narrow-spectrum antibiotics would be reserved for the most serious and intractable infections. New antibiotics would, as a result, be targeting much smaller niche markets (specific pathogens) but would command higher reimbursement rates and therefore provide greater economic incentive for the drug developers to get back in the game.
7. CONCLUDING REMARKS
Clearly since their discovery, antibiotics have had remarkable benefits to mankind in terms of lives saved and suffering abated; however, the indiscriminate use of antibiotics has not been without significant consequences (Figure 1). The 17th century philosopher and scientist, Sir Francis Bacon, said “nature to be commanded, must be obeyed”. That admonishment appears to be particularly relevant to the discovery and development of 21st century medical therapeutics. The current pharmaceutical paradigm for infectious and neoplastic diseases is to administer a drug substance that is more toxic to the target than it is to the patient, thus creating a therapeutic window. For the past 75 years, that strategy has worked for infectious disease management, but as discussed herein, this success has not been without consequences to long-term human health. As an alternative, host tolerance/resistance mechanisms are receiving a great deal of attention and are emerging as a new therapeutic target for the emerging antibiotic resistant pathogens [37], infectious diseases [38,39], as well as cancers [40].
REFERENCES
- Taubes, G. (2008) The bacteria fight back. Science, 321, 356-361. doi:10.1126/science.321.5887.356
- Klevens, R.M., Morrison, M.A., Nadle, J., Petit, S., Gershman, K., Ray, S., et al. (2007) Invasive methicillinresistant Staphylococcus aureus infections in the United States. JAMA, 298, 1763-1771. doi:10.1001/jama.298.15.1763
- Bancroft, E.A. (2007) Antimicrobial resistance: It’s not just for hospitals. JAMA, 298, 1803-1804. doi:10.1001/jama.298.15.1803
- D’Agata, E.M., Webb, G.F., Horn, M.A., Moellering, R.C. and Ruan, S. (2009) Modeling the invasion of community-acquired methicillin-resistant Staphylococcus aureus into hospitals. Clinical Infectious Diseases, 48, 274-284. doi:10.1086/595844
- Farley, J.E. (2008) Epidemiology, clinical manifestations, and treatment options for skin and soft tissue infection caused by community-acquired methicillin-resistant Staphylococcus aureus. Journal of the American Academy of Nurse Practitioners, 20, 85-92. doi:10.1111/j.1745-7599.2007.00290.x
- Wright, G.D. (2010) Antibiotic resistance: Where does it come from and what can we do about it? BMC Biology, 8, 123. doi:10.1186/1741-7007-8-123
- D’Costa V.M., King, C.E., Kalan, L., Morar, M., Sung, W.W., Schwarz, C., et al. (2011) Antibiotic resistance is ancient. Nature, 477, 457-461. doi:10.1038/nature10388
- Bennett, P.M. (2008) Plasmid encoded antibiotic resistance: Acquisition and transfer of antibiotic resistance genes in bacteria. British Journal of Pharmacology, 153, S347-S357. doi:10.1038/sj.bjp.0707607
- Ohnishi, M., Golparian, D., Shimuta, K., Saika, T., Hoshina, S., Iwasaku, K., et al. (2011) Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea? Detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrobial Agents and Chemotherapy, 55, 3538-3545. doi:10.1128/AAC.00325-11
- Dethlefsen, L., Fall-Ngai, M. and Relman, D.A. (2007) An ecological and evolutionary perspective on humanmicrobe mutualism and disease. Nature, 449, 811-818. doi:10.1038/nature06245
- Blaser, M.J. and Kirschner, D. (2007) The equilibria that allow bacterial persistence in human hosts. Nature, 449, 843-849. doi:10.1038/nature06198
- Blaser, M.J. and Falkow, S. (2009) What are the consequences of the disappearing human microbiota? Nature Reviews Microbiology, 7, 887-894. doi:10.1038/nrmicro2245
- Curtis, M.M. and Way, S.S. (2009) Interleukin-17 in host defense against bacterial, mycobacterial and fungal pathogens. Immunology, 126, 177-185. doi:10.1111/j.1365-2567.2008.03017.x
- Annunziato, F., Cosmi, L., Santarlasci, V., Maggi, L., Liotta, F., Mazzinghi, B., et al. (2007) Phenotypic and functional features of human Th17 cells. The Journal of Experimental Medicine, 204, 1849-1861. doi:10.1084/jem.20070663
- Blaser, M.J., Chen, Y. and Reibman, J. (2008) Does Helicobacter pylori protect against asthma and allergy? Gut, 57, 561-567. doi:10.1136/gut.2007.133462
- Koenen, H.J., Smeets, R.L., Vink, P.M., van Rijssen, E., Boots, A.M. and Joosten, I. (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood, 112, 2340-2352. doi:10.1182/blood-2008-01-133967
- Turnbaugh, P.J., Ley, R.E., Hamady, M., Fraser-Liggett, C.M., Knight, R. and Gordon, J.I. (2007) The human microbiome project. Nature, 449, 804-810. doi:10.1038/nature06244
- Jukes, T. (2012) Antibiotics in animal feeds and production. Bioscience, 22, 526-534. doi:10.2307/1296312
- Albrich, W.C., Baughman, W., Schmotzer, B. and Farley, M.M. (2007) Changing characteristics of invasive pneumococcal disease in Metropolitan Atlanta, Georgia, after introduction of a 7-valent pneumococcal conjugate vaccine. Clinical Infectious Diseases, 44, 1569-1576. doi:10.1086/518149
- Regev-Yochay, G., Dagan, R., Raz, M., Carmeli, Y., Shainberg, B., Derazne, E., et al. (2004) Association between carriage of Streptococcus pneumoniae and Staphylococcus aureus in children. JAMA, 292, 716-720. doi:10.1001/jama.292.6.716
- Regev-Yochay, G., Bogaert, D., Malley, R., Hermans, P.W., Veenhoven, R.H., Sanders, E.A., et al. (2008) Does pneumococcal conjugate vaccine influence Staphylococcus aureus carriage in children? Clinical Infectious Diseases, 47, 289-291. doi:10.1086/589573
- Rasko, D.A., Moreira, C.G., Li, D.R., Reading, N.C., Ritchie, J.M., Waldor, M.K., et al. (2008) Targeting QseC signaling and virulence for antibiotic development. Science, 321, 1078-1080. doi:10.1126/science.1160354
- Schneider, D.S. and Ayres, J.S. (2008) Two ways to survive infection: What resistance and tolerance can teach us about treating infectious diseases. Nature Reviews Immunology, 8, 889-895. doi:10.1038/nri2432
- Lamkanfi, M. (2011) Emerging inflammasome effector mechanisms. Nature Reviews Immunology, 11, 213-220. doi:10.1038/nri2936
- Chu, J., Thomas, L.M., Watkins, S.C., Franchi, L., Nunez, G. and Salter, R.D. (2009) Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. Journal of Leukocyte Biology, 86, 1227-1238. doi:10.1189/jlb.0309164
- Dostert, C., Guarda, G., Romero, J.F., Menu, P., Gross, O., Tardivel, A., et al. (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. The Public Library of Science One, 9, e6510.
- Gurcel, L., Abrami, L., Girardin, S., Tschopp, J., van der Goot, F.G. (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell, 126, 1135-1145. doi:10.1016/j.cell.2006.07.033
- Thomas, P.G., Dash, P., Aldridge, J.R., Ellebedy, A.H., Reynolds, C., Funk, A.J., et al. (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity, 30, 566-575. doi:10.1016/j.immuni.2009.02.006
- Cassel, S.L., Eisenbarth, S.C., Lyer, S.S., Sadler, J.J., Colegio, O.R., Tephly, L.A., et al. (2008) The NALP3 inflammasome is essential for the development of silicosis. Proceeding of the National Academy Sciences of the USA, 105, 9035-9040. doi:10.1073/pnas.0803933105
- Dostert, C., Petrilli, V., van Bruggen, R., Steele, C., Mossman, B.T. and Tschopp, J. (2008) Innate immune activetion through NALP3 inflammasome sensing of asbestos and silica. Science, 320, 674-677. doi:10.1126/science.1156995
- Hornung, V., Bauernfeind, F., Halle, A., Samstad, E.O., Kono, H., Rock, K.L., et al. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology, 9, 847- 856. doi:10.1038/ni.1631
- Gasse, P., Riteau, N., Charron, S., Girre, S., Fick, L., Petrilli, V., et al. (2009) Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. American Journal of Respiratory and Critical Care Medicine, 179, 903-913. doi:10.1164/rccm.200808-1274OC
- Mariathasan, S., Weiss, D.S., Newton, K., McBride, J., O’Rourke, K., Roose-Girma, M., et al. (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature, 440, 228-232. doi:10.1038/nature04515
- Salminen, A., Ojala, J., Suuronen, T., Kaarniranta, K. and Kauppinen, A. (2008) Amyloid-β oligomers set fire to inflammasomes and induce Alzheimer’s pathology. Journal of Cellular and Molecular Medicine, 12, 2255-2262. doi:10.1111/j.1582-4934.2008.00496.x
- Yamasaki, K., Muto, J., Taylor, K.R., Cogen, A.L., Audish, D., Bertin, J., et al. (2009) NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. The Journal of Biological Chemistry, 284, 12762-12771. doi:10.1074/jbc.M806084200
- Elinav, E., Strowig, T., Kau, A.L., Henao-Mejia, J., Thaiss, C.A., Booth, C.J., et al. (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell, 145, 745-757. doi:10.1016/j.cell.2011.04.022
- Thacker, J.D., Sassi-Gaha, S., Stephens, C., Purohit, M., Rest, R.F. and Artlett, C.M. (2012) NLRP3 inflammasome is a target for development of broad-spectrum anti-infective drugs. Antimicrobial Agents Chemotherapy, 56, 1921-1930. doi:10.1128/AAC.06372-11
- Henan-Mejia, I., Elinav, E., Strowig, T. and Flavell, R.A. (2012) Inflammasomes: Far beyond inflammation. Nature Immunology, 13, 321-324. doi:10.1038/ni.2257
- Franchi, L., Muñoz-Planillo, R. and Núñez, G. (2012) Sensing and reacting to microbes through inflammasomes. Nature Immunology, 13, 325-332. doi:10.1038/ni.2231
- Zitvogel, L., Kepp, O., Galluzzi, L. and Kroemer, G. (2012) Inflammasomes in carcinogenesis and anticancer immune responses. Nature Immunology, 13, 343-351. doi:10.1038/ni.2224