Open Journal of Genetics
Vol.3 No.1(2013), Article ID:28998,5 pages DOI:10.4236/ojgen.2013.31001
Identification of N-acetylglucosaminyltranferase-IV as a modifier of Epstein-Barr virus BZLF1 activity
![]()
Department of Biology, University of North Carolina Greensboro, Greensboro, USA
Email: aladamso@uncg.edu
Received 22 January 2013; revised 24 February 2013; accepted 2 March 2013
Keywords: Epstein-Barr Virus; BZLF1; Drosophila; CG9384; Gnt-IVb
ABSTRACT
Epstein-Barr virus is a prevalent human herpesvirus, with about 95% of the world’s adult population positive for anti-EBV antigen antibodies. After the initial infection and production of new virus particles, the virus may enter a latent state within a subset of cells, and therefore can remain within the host indefinitely. Epstein-Barr virus contributes to a variety of diseases, including many types of cancers. We have created a model system in Drosophila melanogaster to study the effect of expression of the Epstein-Barr virus protein BZLF1, and to identify cellular proteins that mediate BZLF1 activity. Here we present the results of a genetic screen that determined that the Drosophila melanogaster CG9384 gene (an N-acetylglucosaminyltransferase) is a significant modulator of BZLF1 activity and EBV early lytic replication.
1. INTRODUCTION
Epstein-Barr virus (EBV) is a widespread human herpesvirus, as nearly 95% of the world’s human adult population are positive for anti-EBV antigen antibodies [1]. EBV was discovered in 1957 by Denis Burkitt and Anthony Epstein in tumor samples from Burkitt’s lymphoma [2]. Since then, EBV has been found in association with many cancers, including nasopharyngeal carcinoma, breast cancer, gastric carcinoma, as well as many types of lymphomas, including post-transplant lymphoprolifirative disease, Hodgkin’s lymphoma, and T-cell lymphomas [1]. As EBV is so prevalent, and is associated with such a variety of cancers, it is important to understand how the virus contributes to pathogenesis and carcinogenesis.
EBV replication is controlled by two different types of life cycles: lytic, which occurs upon infection of cells and leads to the production of infectious viral particles, and latent, which is the dormant phase in which the virus produces minimal viral proteins to maintain the viral genomes within immortalized B cells. Lytic replication, which occurs in epithelial cells as well as in B cells (before latency ensues) is broken down into three distinct phases of viral protein production: immediate-early, early and late. The immediate-early (IE) genes, BZLF1 and BRLF1, act like switches to activate lytic replication. The IE proteins are transcription factors that turn on the EBV early genes (which act to replicate the viral genome). The late genes encode virion structural elements [2].
BZLF1 (Z) and BRLF1 (R) not only bind to and transactivate early gene promoters, but they also modulate the host’s intracellular environment by interacting with and altering the activities of many cellular proteins. For example, Z has been shown to interact with the histone acetylase CBP, the transcription factors CREB and Pax5, and the promyelocytic leukemia protein PML [3-5]. Such interactions benefit viral replication, but can be detrimental to the cell.
We previously created a model genetic system to screen for and identify host proteins that interact with Z. We expressed Z in Drosophila eyes, via the GMR system (Glass-mediated response; Glass is an eye-specific transcription factor in Drosophila) [6], which produced a significant mutant phenotype [5]. We performed candidate gene genetic screens, which involved crossing our GMR-Z flies to flies with known mutations in specific genes (such as tumor suppressors), and found some very interesting interactions [5,7]. These interactions have proven to impact EBV lytic replication in human cells [5] (and unpublished observations).
Here, we have performed a random genetic screen by mutagenizing flies and crossing them to GMR-Z flies. We isolated several mutant fly lines that modified the GMR-Z mutant phenotype. One of these modifiers, AJ101, which enhanced the GMR-Z phenotype, was characterized and determined to be a homolog of human Gnt-IVb.
Gnt-IVb is an N-acetylglucosaminyltransferase, which participates in sugar-chain branch formation on proteins that move through the secretory pathway [8,9]. Interestingly, human Gnt-IVb has been found to be misexpressed in several types of cancers, including pancreatic cancer, colorectal carcinoma, and renal cancer [8,10-13]. We found that overexpression of human Gnt-IVb in a human gastric carcinoma cell line enhanced EBV early lytic replication. Therefore, expression levels of Gnt-IVb impact EBV replication and this may indirectly promote carcinogenesis.
2. MATERIALS AND METHODS
2.1. Genetic Screen
iso e males (of a stock with isogenized third chromosomes marked with ebony) were placed in vials containing Whatman paper saturated with 25 mM ethylmethanesulfonate (EMS) in 1% dextrose for 12 hours. These males were crossed to virgin weak GMR-Z females. Male progeny were scored for modification of the weak GMR-Z phenotype.
2.2. Mapping
Recombination mapping: AJ101 was mapped against the third chromosome P element stocks #13230, 14984, 13372, 14258, 13115, 13114, 15252, 16532, 12803, 13126, 12824, 12868, 12629, 12694, 12719 from the Bloomington Stock Center. The lowest level of recombination was found between AJ101 and stock 13115 (at position 70A8). Deficiency mapping: AJ101 was crossed to 18 overlapping deficiencies spanning the region 69A to 77B. All stocks were acquired from the Bloomington Stock Center. AJ101 failed to complement the stock #3126 (with breakpoints 70D2-3; 71E4-5), as well as the smaller deficiency 8074 (70F4-71E1); all other deficiencies in the area complemented AJ101, narrowing the region of AJ101 to 70F. The available mutant alleles for the genes within this region were tested, including Trl (stocks 12088, 13334), CG9384 (stock 22582), mop (stock 15817), bmm (stocks 25926, 15828), CG13472 (stocks 12790, 15978), gnu (stock 3321), and CG17839 (stocks 19525, 22869). AJ101 failed to complement the CG9384 mutant allele only.
2.3. Determination of Heterozygous versus Homozygous AJ101 Larvae
The AJ101 chromosome was balanced over a GFPmarked balancer chromosome, creating the stock AJ101/ TM3 GFP Ser. Adults of this stock were allowed to lay embryos on molasses plates, and the plates examined under a fluorescent dissecting microscope to separate GFP-expressing embryos (heterozygotes) and non-GFP embryos (homozygotes). Separated embryos were allowed to develop into third instar larvae.
2.4. Sequencing
Genomic DNA was isolated from homozygous AJ101 larvae. Briefly, larvae were squished in 50 μL of buffer SB (10 mM Tris pH 8.2, 1 mM EDTA pH 8.0, 25 mM NaCl, 200 μg/ml proteinase K), incubated at 25˚C for 30 min, then incubated at 95˚C for 2 min 5 μL of the lysate was added to PCR reactions (GoTaq, Promega) with three sets of primers from the CG9384 gene sequence (set 1: 5’CAGCGCCTGCATTAGTCATA3’, 5’GTTTCGTTCT GCTCCTCGTC3’; set 2: 5’GTAAGGGTGGCAGTGCA AAT3’, 5’TTTCCTTTCAGCGACGAACT- 3’; set 3: 5’A GTCCAAGGTTTGTCGCACG3’, 5’GCTACCACGAC TGCATCTCA3’). DNA was isolated from an agarose gel with the GeneClean III kit (Q-BIOgene), according to manufacturer’s directions. The primers used for PCR were also used for sequencing the three overlapping gene segments.
2.5. Cell Lines and Transfection
AGS-BDneo (gift of Lindsey Hutt-Fletcher) is an EBVpositive gastric carcinoma cell line and was maintained in Ham’s F12 medium supplemented with 10% fetal bovine serum, penicillin, streptomycin, fungicide, and 500 μg/ml G418. Cells were transfected with either vector alone or a Gnt-IVb-expressing vector (OriGene SC110257) with FuGene HD transfection reagent (Promega), as per the manufacturer’s instructions. 24 hr. post-transfection, cells were either left uninduced, or were induced into lytic replication by the addition of 20 ng/ml TPA and 3 mM sodium butyrate.
2.6. Flow Cytometry
Cells were removed from their culture dishes, washed with PBS, and fixed with 60% acetone in PBS for 10 min. at 4˚C. Cells were washed with PBS/0.5% BSA, and incubated in anti-BMRF1 antibody (Capricorn), diluted 1:200 in Incubation mix (0.3% BSA, 5% goat serum, 0.1% Triton X in PBS) for 1 hr. at room temperature. Cells were washed with PBS/0.5% BSA, and incubated in donkey-anti-mouse-DyLight 488 secondary antibody (Jackson Immunoresearch), diluted 1:400 in Incubation mix, for 1 hr. at room temperature. Cells were washed with PBS/0.5% BSA and resuspended in PBS. Flow cytometry was performed with a Guava easyCyte flow cytometer.
3. RESULTS
3.1. A Random Genetic Screen in Drosophila Identified an Enhancer of the weak GMR-Z Phenotype
To find genetic modifiers of Z, Drosophila iso e male flies were mutagenized with 25 mM ethylmethanesulfonate (EMS) and crossed to weak GMR-Z females, as detailed in Material and Methods. Over 10,000 male progeny were examined for modification of the weak GMR-Z phenotype. One of the modifiers identified, which we named AJ101, yielded a significant enhancement of the weak GMR-Z phenotype (Figure 1). This enhancement included a flattening of the ommatidia, yielding a smoother eye cuticle (compare Figures 1(D) to 1(F)), and a complete loss of eye pigment.
While AJ101 heterozygotes had no mutant phenotype on their own, the AJ101 mutation was found to be homozygous lethal (data not shown). AJ101 homozygotes died at the third instar larval stage.
3.2. AJ101 Maps to the Cytological Region 70F4 and to the Drosophila Gene CG9384
To identify the Drosophila gene which corresponded to the AJ101 modifier, we performed recombination mapping. We initially determined that AJ101 resided on the third chromosome, thus we mapped the location of AJ101 against fifteen P element insertions along the third chromosome. This mapping indicated that AJ101 mapped to a location near chromosomal region 70A8 (data not shown). We then crossed AJ101 to eighteen overlapping deficiencies, spanning chromosomal regions 69A to 77B. The lethal AJ101 mutation failed to complement one of these deficiencies, a deficiency with
500× 2000×
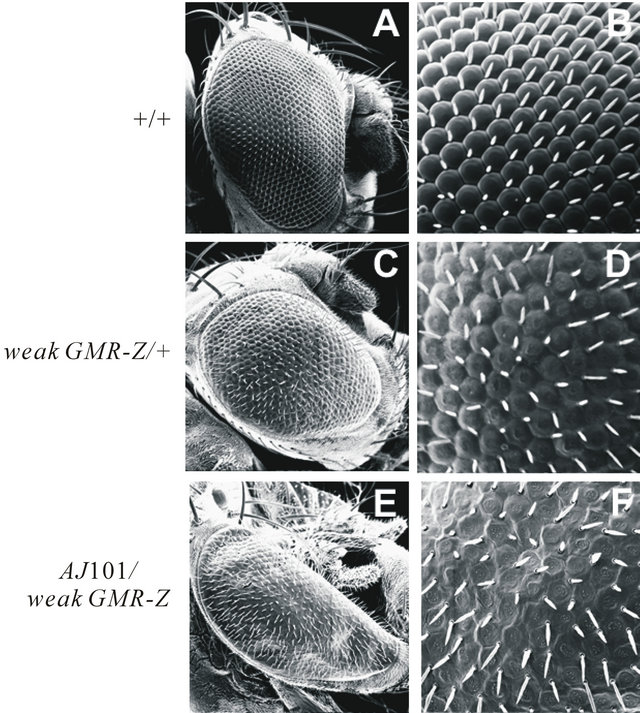
Figure 1. AJ101 is a genetic modifier of weak GMR-Z. A, B: Wild-type. C, D: weak GMR-Z heterozygote. E, F: AJ101/weak GMR-Z trans-heterozygote. SEMs are presented at 500× (A), (C), (E) and 2000× (B), (D), (F).
breakpoints of 70D2-3; 71E4-5 (#3126; see Figure 2). Crosses with smaller deficiencies within these breakpoints narrowed the location of AJ101 to 70F (including #8073, 6551, 8074, 8075, and 24945; Figure 2). Within this area were ten genes for consideration [14]. AJ101 was crossed to available mutant alleles for these genes (Figure 2), and failed to complement a mutation in CG9384. Specifically, we crossed AJ101 to the CG9384 mutant fly line CG9384EY22766/TM3 Sb1 Ser1 (Bloomington stock number 22582). The mutation in this line is due to a P element insertion inserted within the gene, which disrupts the upstream untranslated region at the 5’ end of the mRNA [14]. The resulting AJ101/CG9384EY22766 trans-heterozygote was lethal at third instar, just as the AJ101 homozygotes were. As AJ101 failed to complement this specific mutation, we confirmed the identity of AJ101 as CG9384.
To identify the genomic alteration that caused the AJ101 mutation, the genomic DNA corresponding to CG9384 was PCR-amplified from AJ101 homozygous larvae and sequenced. A nucleotide mutation was found that would cause a conversion of amino acid 512 from a serine to an asparagine within the CG9384 protein.
3.3. CG9384 Is a Homolog of the Human MGAT4B
CG9384 appears to have one spliced transcript, 1764 nucleotides in length, encoding a 587 amino acid long protein. The transcript contains a 105 nucleotide region that is 70% identical to the human mannosyl (alpha-1,3-) glycoprotein beta-1,4-N-acetylglucosaminyltransferase, isozyme B (also called MGAT4B) [14]. The CG9384 protein is 46% identical (65% with conservative substitutions) across amino acids 120 - 567 to the MGAT4B protein, and contains an N-acetylglucosaminyltransferase-IV (GnT-IV) conserved region from amino acids 99 - 412.
N-acetylglucosaminyltransferases (GnTs) participate

Figure 2. The AJ101 mutation maps to the CG9384 gene. Deletion mapping indicated that the AJ101 mutation was located in the cytogenetic region of 70F. Of the genes in this region (between dotted lines) with available mutant alleles, AJ101 failed to complement CG9384. White boxes represent deletions that complemented AJ101, black boxes represent deletions that were lethal in combination with AJ101. Regions shown may not be to scale.
in sugar-chain branch formation within the Golgi apparatus [8,9]. In vertebrates there are six GnTs, which are designated as GnT-I to -VI; these catalyze the transfer of GlcNAc to the core mannose residues of asparaginelinked sugar chains. GnT-IV specifically catalyzes the transfer of GlcNAc from UDP-GlcNAc to the GlcNAc 1 - 2 Man 1 - 3 arm of core oligosaccharide, and forms a GlcNAc 1 - 4 (GlcNAc 1 - 2) Man 1 - 3 structure on the core oligosaccharide [8].
3.4. Overexpression of Gnt-IVb Enhances EBV Early Lytic Replication in EBV-Positive Gastric Carcinoma Cells
To determine whether altered levels of human Gnt-IVb in a human cell line would impact EBV lytic replication, we overexpressed the human Gnt-IVb gene in AGSBDneo cells (gastric carcinoma cells latently infected with EBV), and assessed the impact upon EBV early lytic replication. The EBV early protein BMRF1 is commonly used as an indicator of EBV early lytic replication (for example [15-17]). Figure 3 shows that when cells overexpressed Gnt-IVb, and were subsequently induced into lytic replication, the cells displayed significantly higher levels of the EBV early lytic protein BMRF1, in a dose-dependent manner.
4. DISCUSSION
We performed a random genetic screen in Drosophila to find cellular modifiers of EBV Z activity, with the hope of finding novel interactors that would perhaps be unexpected (as opposed to, say, nuclear transcription factors).

Figure 3. Overexpression of Gnt-IVb enhances EBV early lytic replication. Flow cytometry was performed on cells that overexpressed vector alone (vec), 0.5 μg Gnt-IVb expression vector (Gnt 0.5), or 1.0μg Gnt-IVb expression vector (Gnt 1.0), either without or with induction into lytic replication (24 hr. posttransfection). Cells were immunostained for the EBV early protein BMRF1. The percent of BMRF1-positive cells is presented. The average of three separate experiments are presented. *= p ≤ 0.05 relative to vec induced.
We identified a mutation, which we called AJ101, that enhanced the GMR-Z mutant phenotype, thus enhanced Z activity within epithelial cells. We successfully mapped the AJ101 mutation to the third Drosophila chromosome at position 70F, and more specifically to the Drosophila CG9384 gene.
The human homolog of CG9384 is MGAT4B, also called Gnt-IVb. Gnt-IVb is a glycosyltransferase that participates in the formation of N-glycan branching structures on glycosylated proteins. Gnt-IV, localized to the Golgi, specifically contributes the 1-4GlcNAc branch to a mannose on the core structure [8]. Alteration of Gnt levels, either reduced or increased, have serious consequences upon a cell, as cell surface and secreted proteins, among others, are glycosylated. This glycosylation is important for proper localization (or secretion) and function of proteins. The alteration of Gnt-IV levels (both the a and b forms) has been linked to cancer and diabetes. Increased Gnt-IVb levels have been found in association with pancreatic and colorectal cancers [10-12], and decreased levels have been found in association with renal cell cancer [13]. Interestingly, Takamatsu et al. found that Gnt-IVb-deficient mice had an increase in Gnt-IVa expression (an apparent compensation mechanism), but still showed abnormalities to hemostasis, including a decreased number of neutrophils and increased lymphocyte cellularity [18].
In relation to EBV Z, which is a nuclear transcription factor that would not pass through the Golgi and therefore would not likely be exposed to Gnt-IVb, the effect of misexpression of Gnt-IVb must be an indirect one. However it is logical to assume that alterations to glycolsylation of proteins (namely cell surface and secreted proteins) would alter those proteins’ functions, and affect numerous signal transduction processes. Z has previously been shown to interact with signal transduction pathways, as well as transcription factors that are activated via signal transduction pathways [3,17] (and unpublished observations). Furthermore, EBV biology in general is dependent upon signal transduction pathways [19-22]. Therefore, alterations to the levels and/or functions of signaling molecules and cell surface receptors would likely affect Z activity and EBV replication in general, as well as affect host cell immune responses. Misexpression of glycosyltransferases, such as Gnt-IVb, has been associated with various cancers, and as an alteration of the Drosophila Gnt-IVb in Z-expressing tissues enhanced Z activity, it follows that misexpression of glycosyl-transferases in tissues that are infected with EBV may lead to increases in EBV replication and perhaps to increases in EBV’s contribution to carcinogenesis.
5. ACKNOWLEDGEMENTS
This work was supported by NIH grants 1R21DE014602-01 and 1R15AI072699-01. We would like to thank Adrienne Jones for isolating the AJ101 mutant, and Dennis LaJeunesse for assistance with the genetic screen.
![]()
![]()
REFERENCES
- Rickinson, A.B. and Kieff, E. (2007) Epstein-Barr virus. In: Knipe, D.M. and Howley, P.M., Eds., Field’s Virology, Lippincott Williams & Wilkins, Philadelphia, 2655- 2700.
- Kieff, E. and Rickinson, A.B. (2007) Epstein-Barr virus and its replication. In: Field’s Virology, Lippincott Williams & Wilkins, Philadelphia, 2603-2654.
- Adamson, A.L. and Kenney, S. (1999) The Epstein-Barr virus BZLF1 protein interacts physically and functionally with the histone acetylase CREB-binding protein. Journal of virology, 73, 6551-6518.
- Bowling, B.L. and Adamson, A.L. (2006) Functional interactions between the Epstein-Barr virus BZLF1 protein and the promyelocytic leukemia protein. Virus Research, 117, 244-253. doi:10.1016/j.virusres.2005.10.018
- Adamson, A.L., Wright, N. and LaJeunesse, D.R. (2005) Modeling early Epstein-Barr virus infection in Drosophila melanogaster: The BZLF1 protein. Genetics, 171, 1125- 1135. doi:10.1534/genetics.105.042572
- Hay, B.A., Wolff, T. and Rubin, G. (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development (Cambridge, England), 120, 2121-2129.
- Adamson, A.L. and LaJeunesse, D. (2012) A study of Epstein-Barr virus BRLF1 activity in a Drosophila model system. TSWJ 2012, 1-9.
- Taniguchi, N. and Korekane, H. (2011) Branched N-glycans and their implications for cell adhesion, signaling and clinical applications for cancer biomarkers and in therapeutics. BMB Reports, 44, 772-781. doi:10.5483/BMBRep.2011.44.12.772
- Gleeson, P.A. and Schachter, H. (1983) Control of glycoprotein synthesis. The Journal of Biological Chemistry, 258, 6162-6173.
- Ide, Y., Miyoshi, E., Nakagawa, T., Gu, J., Tanemura, M., Nishida, T., Ito, T., Yamamoto, H., Kozutsumi, Y. and Taniguchi, N. (2006) Aberrant expression of N-acetylglucosaminyltransferase-IVa and IVb (GnT-IVa and b) in pancreatic cancer. Biochemical and Biophysical Research Communications, 341, 478-482. doi:10.1016/j.bbrc.2005.12.208
- Nan, B.C., Shao, D.M., Chen, H.L., Huang, Y., Gu, J.X., Zhang, Y.B. and Wu, Z.G. (1998) Alteration of N-acetylglucosaminyltransferases in pancreatic carcinoma. Glycoconjugate Journal, 15, 1033-1037. doi:10.1023/A:1006950311937
- D’Arrigo, A., Belluco, C., Ambrosi, A., Digito, M., Esposito, G., Bertola, A., Fabris, M., Nofrate, V., Mammano, E., Leon, A., Nitti, D. and Lise, M. (2005) Metastatic transcriptional pattern revealed by gene expression profiling in primary colorectal carcinoma. International Journal of Cancer, 115, 256-262. doi:10.1002/ijc.20883
- Zhu, T.Y., Chen, H.L., Gu, J.X., Zhang, Y.F., Zhang, Y.K. and Zhang, R.A. (1997) Changes in N-acetylglucosaminyltransferase III, IV and V in renal cell carcinoma. Journal of Cancer Research and Clinical Oncology, 123, 296-299.
- McQuilton, P., St. Pierre, S.E., Thurmond, J. and Consortium, T.F. (2012) FlyBase 101—The basics of navigating FlyBase. Nucleic Acids Research, 40, D706-D714. doi:10.1093/nar/gkr1030
- Calderwood, M.A., Holthaus, A.M. and Johannsen, E. (2008) The Epstein-Barr virus LF2 protein inhibits viral replication. Journal of Virology, 82, 8509-19. doi:10.1128/JVI.00315-08
- Liu, S., Li, H., Chen, L., Yang, L., Li, L., Tao, Y., Li, W., Li, Z., Liu, H., Tang, M., Bode, A.M., Dong, Z. and Cao, Y. (2012) (-)-Epigallocatechin-3-gallate inhibition of Epstein-Barr virus spontaneous lytic infection involves ERK1/2 and PI3-K/Akt signaling in EBV-positive cells. Carcinogenesis.
- Adamson, A.L., Darr, D., Holley-Guthrie, E., Johnson, R.A., Mauser, A., Swenson, J. and Kenney, S. (2000) Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun Nterminal kinases. Journal of Virology, 74, 1224-1233. doi:10.1128/JVI.74.3.1224-1233.2000
- Takamatsu, S., Antonopoulos, A., Ohtsubo, K., Ditto, D., Chiba, Y., Le, D.T., Morris, H.R., Haslam, S.M., Dell, A., Marth, J.D. and Taniguchi, N. (2010) Physiological and glycomic characterization of N-acetylglucosaminyltransferase-IVa and -IVb double deficient mice. Glycobiology, 20, 485-497. doi:10.1093/glycob/cwp200
- Brinkmann, M.M. and Schulz, T.F. (2006) Regulation of intracellular signalling by the terminal membrane proteins of members of the Gammaherpesvirinae. The Journal of General Virology, 87, 1047-1074. doi:10.1099/vir.0.81598-0
- Izumi, K.M. (2004) Epstein-Barr virus signal transduction and B-lymphocyte growth transformation. Progress in Molecular and Subcellular Biology, 36, 269-288.
- Mosialos, G. (2001) Cytokine signaling and Epstein-Barr virus-mediated cell transformation. Cytokine & Growth Factor Reviews, 12, 259-270. doi:10.1016/S1359-6101(00)00035-6
- Bryant, H. and Farrell, P.J. (2002) Signal transduction and transcription factor modification during reactivation of Epstein-Barr virus from latency. Journal of Virology, 76, 10290-10298. doi:10.1128/JVI.76.20.10290-10298.2002