Open Journal of Genetics
Vol.1 No.1(2011), Article ID:5279,8 pages DOI:10.4236/ojgen.2011.11001
Xenogamy and current obesity pandemics
![]()
Department of Evolutionary Immunology, Andent Inc, Jersey City, USA
E-mail: rumyan1@yahoo.com
Received 11 April 2011; revised 30 May 2011; accepted 17 June 2011.
Keywords: Biodiversity; Evolution; Genetics; Heterozygosity; Leptin-Receptor System
ABSTRACT
This article unites the results from an integrative reconsideration of actual data about obesity from the viewpoint of recent developments in general pathology, epidemiology, immunology, genetics, and evolution. The focus is on the origin of genetic diversity in obesity manifestations on different levels of the disease. In contrast to the current paradigm of nutritive origin of obesity, the revealed set of evidences shows that the origin of obesity is based on either hereditary or post-infectious incongruence between the regulators of fat metabolism and their receptors. The incongruence may be created either by the destruction of receptors during infection or by genome mutations, which by natural selection, lead both to life-saving incongruence between the infectious agent and its receptor, and simultaneously, in evolution to a lifethreatening obesogenic incongruence in the regulator-receptor interaction. The pandemic spread of obesity is induced by xenogamous intercourse between constitutionally incongruent ethnoses, which regulator-receptor evolution has processed at different epidemic conditions. The achieved results have allowed a replenish model for the molecular pathogenesis of obesity to explain the genetics of both obesity and its pandemic spread. The formerly maladroit issues of obesity origin, genetic, pathogenesis, and pandemic spread are transformed in an entire replenished system.
1. INTRODUCTION
Obesity presents one of the biggest and catastrophically growing problems in the world’s health. Before the middle of 20th century, the disease was thought to be rare. Over the last half of the century, the incidence of obesity began grow so sharply that in 1997, the World Health Organization formally recognized the disease as a global epidemic. The worldwide obesity rate increased from 2.3% to 19.6% between 1990 and 2000. The United States (US) has succumbed to the largely unrecognized obesity epidemic since 1960. In just two decades, the incidence of adult obesity has increased from 15% in 1980 to over 27% in 2000 [1,2]. An equally large growth rate has been observed in juvenile obesity: from less than 5% in 1970 to over 15% in 1998, a three-fold increase in only 30 years. The proper danger of obesity is forced by its close association with a many harm illnesses, such as heart disease, diabetes, high blood pressure, atherosclerosis, stroke, different kinds of cancer, liver disease, gout, and disease of the joints. Over 300,000 excess adult deaths in the US each year are related to obesity [1]. The epidemic continues to progress in a manner similar to a worldwide pandemic.
The existing system for counteracting obesity epidemic is based on the nutritive paradigm of the disease origin and pathogenesis. According to the paradigm, humankind has become “obesogenic”, thanks to increased food intake, non-healthy foods, and physical inactivity [3]. Healthy choices in nutrition and physical activity are considered most effective in combating obesity [4]. The standard advice to obese individuals is to eat less and exercise more, but this has done nothing to stem the obesity epidemic. Efficacy of the means recommended for obesity prevention and treatment is very disappointing. The situation could change if scientists could decipher the origin of obesity. New insights into origin and pathogenesis of obesity, therefore, are sorely needed. The present article aimed to initiate filling in the gaps of contemporary obesitology by means of an integration of relevant recent achievements of all-pathological, immunogenetic, genetic, molecular, and evolutionary discoveries.
2. MATERIALS AND METHODS
The article presents the results from reconsidering current data regarding epidemiology, clinical manifestations, and molecular pathogenesis of obesity from the viewpoint of recent all-pathological, immunogenetic, genetic, and evolutionary discoveries. The main accent was on the signs of genetic predilection to obesity amongst different human populations, ethnoses, and individuals. Special attention was paid to the observations of genetic peculiarities of different locations of the obese body followed to cellular and molecular levels and to the comprehension of their origin from the viewpoint of xenogamy named also heterozygous fertilization, genetic admixture, xenogenesis, crossbreeding, outbreeding. The terms are broadly used in genetics to describe the union of genetically unrelated organisms within the same species.
3. RESULTS AND DISCUSSION
3.1. Population and Ethnic Diversity in Obesity Prevalence
Obesity affects up to 10% of the adults in the world today. Meanwhile, according to information presented by 32 countries [5], at least a 10-fold variation in obesity incidence rates exists worldwide (Table 1). India, China, Papua New Guinea, Japan, and Malaysia are in the group of five least obesogenic countries. Amongst of the group of most ‘obesogenic’ populations, the highly industrialized US shares the neighborhood with Samoa, Jordan, Cyprus, and Kuwait, whereas France, Vanuatu, New Zealand, Canada, and Australia are in the middle of the discussed list.
These observations are in contrast with the most orthodox postulates about the causes of obesity. At the same time, they reveal initial milestones in the way of deciphering of both the origin of obesity and the genetic components of the disease pathogenesis.
3.2. Pathogenesis of Obesity
In recent years, tremendous advances have been made in the understanding of both the molecular physiology of body fat regulation and the molecular pathogenesis of obesity [6-8]. These achievements created important prerequisites for further development of relevant knowledge not only of obesity pathogenesis, but also of the specificity of genetic of both obesity development and epidemic dispersion of the disease among current human population.
The current knowledge of forces propelling the development of individual obesogenesis is successfully progressed. In contrast to the nutritive hypothesis of obesity origin and pathogenesis at least three principal ways for the induction of obesity may be exploited today. Firstly, obesity can develop as the result of parental genome mutation changing the structure of body fat regulation. Secondly, development of obesity may follow an infection. Thirdly, the disturbance of food intake regulation can arise as a result of xenogamy (heterozygous fertilization, genetic admixture, xenogenesis, crossbreeding, outbreeding) predestined by the evolutionary

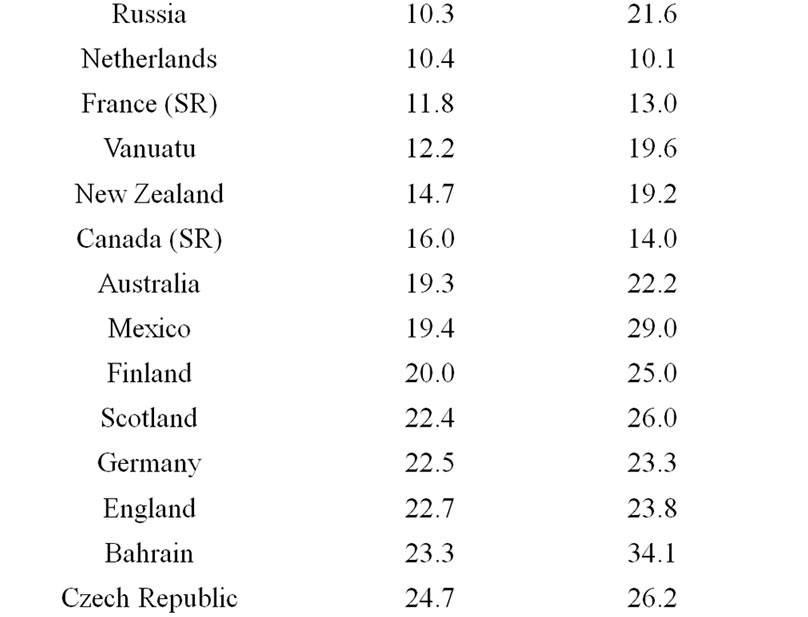
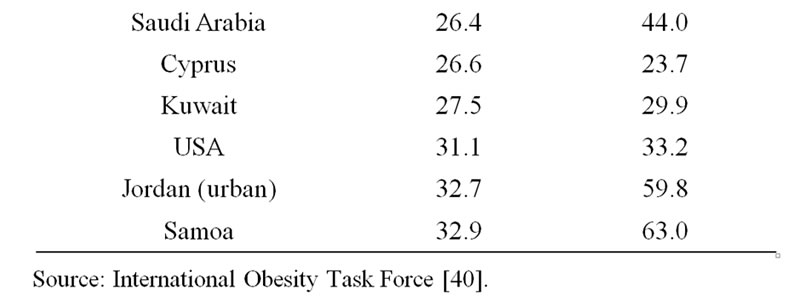
Table 1. Prevalence of Obesity in Several Countries Worldwide.
diversification of parents’ make-up performed through the evolution of ecologically different ethnoses.[9].
3.2.1. Mutative Obesogenesis
Every kind of living being is provided with a constitutive physiological system that maintains normal body structure within its genetically predetermined shape and size. The part of the system that regulates body fat in mammals has now been discovered to the molecular level [8,10]. The control of satiety and the ability to burn fat for energy is performed by a particular set of hormones and other molecular physiological agents. The hormonal regulation of food intake is mainly performed by leptin and insulin [6-8]. They stimulate us to eat more when fat storages are low and less when fat storages are high.
Leptin is produced peripherally by fat storages in adipose tissue and controls the appetite through its actions on relevant hypothalamic cells provided with special leptin receptors. The inhibition of excess food intake is initiated by leptin production performed by satiated adipose tissue, continued by leptin interaction with its hypothalamic receptors, and results in the suppressive response of the hypothalamus addressed to the fat-storehouses [8,10]. The disturbance of any of these links leads the adipose tissue to become unsatiated and begin an uncontrollable intake of foodstuff. It has been shown that the mutant change of the structure and function of leptin leads to the development of obesity [11].
Mutative disturbances caused by sporadic mutant modification of either leptin or its receptor may influence regulatory interaction between leptin and its receptor [11,12]. The same effect may be performed by the mutant absence of any of the coactants. In any case, the disturbances lead to structural resistance (constitutional immunity) of the hypothalamus to the signals of the body’s satiety and to its inability to inhibit excess food intake. As a result, the diseased body does not become satisfied and “makes a pig of oneself”, allowing the development of obesity.
Mutant inadequacy in the production of leptin can function on the level of adipose cells that leads to congenital leptin deficiency, resulting in an insatiable appetite and severe early-onset obesity with numerous other clinical abnormalities; all of which leptin treatment can correct [11,12]. The same result could be achieved by mutations leading to a change of leptin structure. Leptin treatment can correct both state [12]. It should be noted that the administration of leptin might be effective only in a small subset of obese individuals who are leptin deficient. It has been shown that subjects who are heterozygous for the leptin mutation also develop obesity but not as severe as in homozygotes [11].
The mutations in the formation of leptin receptors disturb the discussed regulatory function on the level of hypothalamus. Leptin treatment cannot correct the blocking effect of the disturbance. Most obese individuals have been found to have high levels of leptin and are thought to be leptin resistant [8]. Thus, in some obese bodies, the leptin function can be normal or its content increased by leptin treatment, but its performance can be stifled by a derangement of receptor cells, that is, by the body’s immunity to leptin arising because of incongruent receptors [12,13]. This state of hypothalamus immunity to leptin is thought to explain why the administration of leptin has not been shown to be effective in suppressing appetite in most obese people [6].
An analogous mutant decrease in the sensitivity of cells may prevent the specific action of physiological agents (hormones, enzymes, mediators, chalones, and so on). Constitutional immunity of cells to insulin is a major determinant in the decline of glucose tolerance. The non-insulin-dependent (type II) diabetes mellitus is characterized by hyperglycemia in the presence of higher than normal levels of plasma-insulin. Similarly, an immunopathogenic decrease in cell sensitivity to vitamin D3 determines the familiar forms of rachitis. Constitutional immunity of cells to androgenes causes the phenomenon of testicular feminization. The analogous genetic resistance of cells to corticosteroids determines the pathogenesis of Cushing disease [14].
3.2.2. Post-Infectious Obesogenesis
Although the spread of widely dispersed cases of obesity is progressing as a worldwide pandemic, the possibility that infections might be etiologic agents in obesity has currently received relatively little confirmation. Animal experiments link obesity to four animal viruses (rabies virus, Rous-associated virus, Borna virus, and SMAM-1). Only one human virus, adenovirus 36 (AD36), has been shown to increase adiposity in experimentally infected chickens, mice, and marmosets. Virus-induced obesity in mice alters their response to leptin [15] that act on the thalamus cells. On the other hand the attempts have been made in the past allowed the adenovirus infection alters lipid metabolisms in animal adipocytes too [16]. But a direct correlation of human obesity with an epidemic of any infection has not been found for a long time [11,18-22]. The situation began to change after it had been argued that obesogenic function does not belong to infection by itself but to a constitutional post-infectious immunity induced by it [34].
Direct relationship of a child’s obesity with their previous infection with AD36 has recently been confirmed. The bodies of 124 children were searched for traces of previous AD36 infection—the presence specific antibodies against this virus. Nineteen children revealed specific antiviral antibodies, the responsive signs of past AD36 infection (15%). The majority of children found to be AD36-positive were obese (15 (78%) of 19 children). The responsive signs of foregoing infection, AD36 antibodies, were significantly more frequent in obese children (15 (22%) of 67 children) than in non-obese children (4 (7%) of 57 children) [23].
The studies demonstrated a link between AD36 and obesity, but did not deliver the evidence of how the virus itself causes people to become obese because a long time had passed between the infection and the obesity manifestation. By the time a person becomes obese, the virus is long gone and the person is no longer infectious. Next was an attempt to answer the question from the viewpoint of the nature of constitutional post-infectious immunity [24,25]. The approach was based on the discovery of an unexpected commonality in the structures and functions of molecular physiological and infectious agents.
Tetanus toxin interacts with the thyroid thyrotropin receptor. Its binding can be inhibited by thyrotropin [26]. The rabies virus receptor appears to be closely associated with the acetylcholine reception [27]. The EpsteinBarr virus has a strong tropism for lymphocytes and was thought those of the complement C3 receptor [28]. The human T-cell leukemia virus preferentially infects cells of the helper/inducer class [29]. The cardioviruses and influenza A and B viruses bind to the glycophorin A molecule on human erythrocytes, and this glycoprotein also carries the M and N blood group antigens [30]. The B chain of cholera toxin and the beta-subunits of thyrotropin, luteinizing hormone, human chorionic gonadotropin, and follicle stimulating hormone are shown to have a region of sequence analogy believed to correlate with their ability to bind to the receptor on cell membranes. A sequence analogy is also defined in the alpha-subunits of these glycoprotein hormones and in the region of the cholera toxin A1 chain responsible for adenylate cyclase activation [31].
Following the data, it was permissible to suppose that in fact, all “viral” and “bacterial” receptors have initially been evolved to provide physiological functions of hormones or other physiologically active molecules [9,25]. Evolutionary adaptation of relevant microbes to a parasitic way of life could be performed using pre-existent hormonal receptors.
A constitutional component of post-infectious immunity is formed during a relevant infectious process by the annihilation of susceptible cell structures, including relevant receptors. After recovering, the individual becomes constitutionally immune against re-infection [24,25]. In the discussed case of proposed exact sameness of a receptor both for the AD36 virus and leptin, the formation of post-infectious immunity against infection means a simultaneous formation of both hypothalamus immunity to leptin and its inability for the signaling of adipose tissue regarding excess food intake. The prevalence of post-infectious obesogenesis depends on the intensity of relevant infectious epidemic, that is, the number of susceptible people existent in the observed population [25].
The origin of cell immunity to specific molecular physiological agents is analogous to those in constitutional immunity to infections. In a case of molecular ecological aggression of harmful infectious agents, the specific features of the molecular constitution of the attacked body provide constitutional immunity. The success or failure of microbial aggression also depends on the precise molecular complementariness of the infectious agents with the molecular make-up of the attacked organism, namely its cellular, subcellular, and molecular structures [25].
The organisms possessing this modified molecular constitution are insusceptible to the relevant infectious agent. They do not contain susceptible cellular, subcellular, and molecular structures, whereas organisms in which the number of constitutionally susceptible structures is high, the infectious process develops in a very severe form. Their chances for everlasting life by means of self-reproduction are diminished sharply. Corresponding microbes do not affect those organisms, which are able to produce genetically immune offspring, while susceptible individuals of the same species become ill and die without progeny [32]. On repeated exposures of a population to a given pathogen, the progeny of immune mutants comes, in evolution, to predominate in the population; an individual protective mutation becomes the property of a group, then of a population, ethnoses, and finally of the species. The beneficial attributes of constitutional immunity to infectious agents are propagated in the population thanks to natural selection as an adaptation to life-threatening environmental agents. The selection for the life-saving modification of viral receptors means the simultaneous formation of the life-threatening disturbance of the leptin reception.
3.2.3. Xenogamous Obesogenesis
Humans are very diverse in their predisposition to obesity, as well as in the grade of its expression [9,25]. The forms and rates of obesity progression may be different not only in various individuals, but also in different organs. Equally with the total affection of almost all the body, there may be several areas of restricted fat locations around the body (Figure 1).
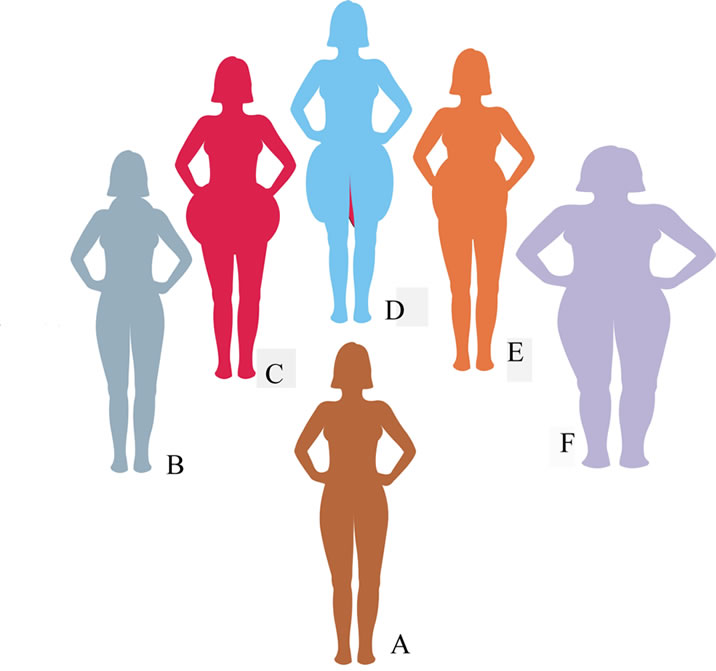
Figure 1. Some variants of the size and location of obese zones around human body (38).
In most cases, obesity is presented by local manifestations of individually different grades. Their sizes vary from solitary small lipomas to multiple massive bulges forming abnormal enlargements of involved parts of the body. The local areas of a human body where obesity can be observed both on women and men include the facial jowls, neck, upper back of the arms, upper back underneath the bra straps, areas near the underarms, waist, love handles, hips, breast, buttocks, abdomen, belly, thighs, knees, calves, and inner organs as well [34]. Any local fat-storehouse is a growth of fat cell population surrounded by a thin, fibrous capsule.
Local character of affections is a very important feature of obesity, the origin of which has been ignored by previous pathogenetic investigations [9,25]. Meanwhile, its observations allow that physiological functions of fat metabolism and its regulations in obesity are disturbed differently in different body areas of the same person. It can also be concluded that within every obese human body, there are at least two co-existing clones of fat cells with the opposite predisposition to obesity and two distinct systems for regulation of food intake and fat metabolism. One of the clones is predisposed to the development of obesity. The size of obese cell populations and their dislocation around the body predestine individual diversity in the course and severity of obesity.
Despite the vastness of differences between existing diseases, one striking similarity unites all of them without exception. This similarity is seen in the mosaic locations and sizes of the lesions [32,35]. Mosaicism of a diseased state is the condition in which the phenomenon genetically determines the variations in its manifestations. Mosaicism of affections is a syndrome universal for any kind of pathology. All diseases (heart, circulatory, pulmonary, mental, skeletal, renal, gastrointestinal, endocrine, or metabolic) are characteristic of the diverse grade and focal dislocation of specific lesions. The existence of this phenomenon was obvious for the general paradigm of pathology, but its origin and pathogenetic significance were only discovered at the end of the 20th century [32,36].
The dapple dispersion of infectious damages is also caused by intra-individual diversity in the local sensitivity of some parts of outwardly homogenous tissue to the action of relevant pathogen. How can we account for the fact that the identical cells of an organism differ from one another when they all contain the same genome complex? Up to the end of the 20th century, it was a riddle for both pathology and genetics [32,36]. Recent advances in the discovery of hereditary immunity [25], especially of its constitutional mechanisms, allowed the understanding of the origin of this phenomenon.
The existence of local intraindividual differences in susceptibility/resistance to infection explains why a given agent affects some parts of a tissue, while other morphologically identical components of the same organism remain uninvolved, although both exist under the same conditions and may be equally influenced by the agent. According to data from many experiments and observations, the mating of resistant and susceptible individuals gives rise to progeny with intermediate degrees of infectious lesions [32,37-40]. The spotty disposition of obesity manifestations can also be considered as an example of same universal law of pathology.
Modern humankind is characteristic of the hugeness of racial and ethnic differences formed in evolution. Anatomically, modern humans emerged in one place, probably northern Africa’s savannah over 200,000 years ago [41]. But the evolution of humankind was not finished at that time. Nearly 60,000 years ago, different groups of early Homo sapiens began to sweep out of Africa and disperse around distinct parts of other continents. Their wanderings around the world substantially expanded both the quantity and quality of the evolutionary driving forces they encountered on their ways. Inhabiting ecologically disparate geographical areas, migrants continued to evolve independently into five anatomically different races and a multiplicity of segregated ethnic groups. Nearly all differences between current human races, ethnoses, and populations are due to events that occurred outside of Africa [42]. The subsequent genetic admixture between distinct human populations increased the number of new traits in human constitution, while many thousands of wandering generations were subjected to various selective pressures acted on by the plethora of unprecedentedly distinct physical, chemical, and bioecological agents. Interethnic differences in the regulation of fat metabolism should be subjected to the same kind of genesis.
These genetically predetermined differences are proof of heterozygosity: one parent passes a lack of receptivity to relevant regulators in some parts of the descendant body on to the descendant, while unaffected parts are inherited from the second parent. This analogy allows us to suppose the same genetic origin in the case of the intraindividual diversity in obesity. According to epidemiological and clinical observations, xenogamy functions as an important co-maker of obesity genetics and its variable manifestations [9,25]. The cross-breeding between healthy persons possessed different structures for the regulation of fat metabolism, giving rise to obese progeny with different degrees of the disease.
The view of heterozygous origin of local obesogenesis can explain why the disease affects some parts of a body, while other parts of the same organism remain uninvolved, although both exist under the same conditions. The loci of affections appear as encapsulated bulges, and the distribution of such foci are distinct for different people and for different parts of the same person. The heterozygous body of the descendant can contain at least either a set of two distinct clones of adipose tissue or two distinct regulators of fat metabolism.
3.3. The Current Spurt in Obesity Incidence
The application of the three above schemes of obesity pathogenesis can help to explain the leading propelling causes of current epidemic progression of obesity prevalence. According to the above performed epidemiological and pathogenetic analysis, the obesogenic functions of mutations undoubtedly possess important roles in the pathogenesis of relevant forms of the disease. Regretfully, none of the mutations by themselves are able to explain the pandemic spread of obesity. Neither somatic nor genomic obesogenic mutations could be widely disseminated in humankind thanks to their rarity, randomness, and to the counteraction of natural selection. Thus, the undoubted existence of mutative obesogenesis cannot be used for the explanation of the moving forces of the pandemic spread of obesity.
In contrast, post-infectious obesogenesis may have more distribution among most modern populations. The possibility of the progressive spread of post-infectious obesity depends on the intensity of the relevant obesogenic infectious epidemic and on the efficacy of its antiinfectious prophylaxis. Industrialized societies possess the greatest potencies for the prevention of infectious epidemics. Undoubtedly, they get more chances for the restriction of post-infectious obesogenesis among their citizens.
The potency of xenogamous obesogenesis has very long history. Although obesity is named as a disease of civilization, it is not a recent phenomenon. The natural history of obesity can be traced back at least 25,000 years ago thanks to sculptures of corpulent women made by stone-age artists, such as the “Venus of Willendorf” found in several sites around the world. But potentially obesogenic forces could have begun to function among humankind far earlier. Infectious drivers of obesogenesis emerged at the savannah stage of human descent (1.8 million years ago), when predecessors of modern humans met the plethora of life-threatening infectious agents for the first time [25].
The exodus out of North East Africa and subsequent dispersion around the world over the last 60,000 years has resulted in a wide biological diversification of the species and a strong self-segregation of its tribes from each other. Some tribes moved back to tropical South Africa, the homeland of their predecessors. Other groups migrated in the Euro-Asian or South-Asian ways. Their further evolution was performed by the forces, which propelled biological and social diversification of the species over its dispersion around the world. Inhabiting ecologically disparate geographical areas, migrants continued to evolve independently into five anatomically different races and a multiplicity of segregated ethnic groups. These new ways of life did not favor a xenogamous epidemic spread of obesity, except when segregation was broken forcedly, for instance, by aggressive tribes. The influence of xenogamy on the distribution of obesity among the members of separated ethnic groups was restricted.
Today, the situation is becoming the opposite. Thanks to growing industrialization, urbanization, globalization, and migration, most urban populations are ethnically mixed. The genomes of modern urbanized humans become the mosaics composed of genetic segments inherited from a long row of ethnically different ancestors. The spread of obesity became pandemic, intensified by the growing expansion of xenogamy, the reproductive intercourses between ethnoses, which proceeded at different environmental conditions for previous evolution. The currently observed increasing incidence of obesity, as well as many other diseases, depends on the intensity of the population’s genetic admixture promoted within ethnically mixed populations.
More mixed African Americans are more likely to suffer from obesity than any other racial or ethnic population. [43,44] Obesity data shows African Americans have a 51% higher prevalence of obesity and Hispanics have 21% higher obesity prevalence compared with whites [45]. This kind of pathology is now more characteristic of any mixed population. The current pandemic spread of obesity is intensified today by the growing expansion of xenogamy.
4. CONCLUSIONS
The above-presented results of the reconsideration of the actual data regarding obesity from the viewpoint of recent all-pathological, epidemiological, immunological, genetic, and evolutionary discoveries allowed a new integrative paradigm about the origin and pandemic spread of the disease to be formed. The study has been performed by exposure and analysis of various epidemiological, clinical, immunological, genetic, and experimental data concerning principal characteristics common for both obesity and other kinds of diseases, especially of infectious ones. This approach allowed us to expose and highlight new ways toward the molecular level of immunogenic and genetic factors involved in the appearance, evolution, spreading, and maintenance of obesity.
Seven decisive factors participated in the creation of obesity: (1) Appropriate infection; (2) Constitutional post-infectious immunity; (3) Epidemics of the infection; (4) Natural selection for hereditary immunity against the infection; (5) Ethnic diversification of humankind; (6) Inter-ethnic crossbreeding; and (7) Globalization of humankind. Points 1 to 4 predestined the origin of the disease, whereas points 5 to 7 formed prerequisites and propelling forces for its pandemic spread.
The revealed set of evidences allows for the demand that the origin of obesity is based on the exact sameness of a receptor both for the relevant virus and leptin and on the formation of either post-infectious or hereditary immunity to leptin, a physiological regulator of food intake. Most cases of resistance exist as constitutional incongruence between leptin and its receptors. This immunity is created either by elimination of leptin receptors during virus infection or by natural selection of the life-saving mutant incongruence between a virus and its receptor that lead simultaneously, in evolution, to life-threatening incongruence in hormone-receptor co-action.
Like any other disease, obesity is characteristic of diversity in the course, manifestations, and severity of specific affections, and their sizes and stochastic focal disposition around the body. Individual differences in the manifestations and severity of discussed disease are associated with the phenomenon of stochastic focal distribution of obesity zones around a body. The differences are of genetic origin. This phenomenon is analogous to those characteristics of any other kind of pathology, being explained by the hybridization of persons possessing different grades of genetic predisposition to relevant pathogens.
The methodological approach used in the performed study allowed us to present the first genetic explanation for the modern increase in obesity incidence. Like any other hormonal disturbance, obesity could arise as a result of constitutional incongruence between relevant hormones and their receptors. The obesogenic molecular make-up could arise and spread among the worldwide population because of both long-time natural selection for genetic immunity to relevant infection and subsequent xenogamy-crossbreeding among mutually distinct parents. From this point of view, life-threatening obesity could be considered as a reckoning, both for the lifesaving evolution of beneficial genetic immunity to relevant infection and for the production of offspring unlike their parents.
The integrative view of the origin of obesity and its pandemic spread supplies a framework for understanding the genetic nature of obesity pandemic and its rising incidence in the current worldwide population. The new paradigm allows us to explain the origin of obesity and its pandemics without McDonald’s, Game Boys, and so on. It also forces us to reconsider the perspective of future investigations and to reassess the principles for obesity prevention and healing.
REFERENCES
- Flegal K.M., Carroll M.D., Kuczmarski R.J., et al. (1998) Overweight and obesity in the United States: prevalence and trends, 1960-1994. International Journal of Obesity and Related Metabolic Disorders, 22, 39-47. doi:10.1038/sj.ijo.0800541
- Mokdad A.H., Ford E.S., Vinicor F., et al. (2001) The continuing epidemics of obesity and diabetes in the United States. The Journal of the American Medical Association, 286, 1195-1200. doi:10.1001/jama.286.10.1195
- Habbu A., Lakkis N.M. and Dokainish H. (2006) The obesity paradox: Fact or fiction? The American Journal of Cardiology, 98, 944-948. doi:10.1016/j.amjcard.2006.04.039
- CDC. (2010) Overweight and obesity. Centers for Diseases Control and Prevention.
- International Obesity Task Force (2009) The Australian and New Zealand obesity society. Prevalence of Obesity in Several Countries Worldwide.
- Flier J.S. (2004) Obesity wars: Molecular progress confronts an expanding epidemic. Cell, 116, 337-350. doi:10.1016/S0092-8674(03)01081-X
- Friedman J.M. and Halaas J.L. (1998) Leptin and the regulation of body weight in mammals. Nature, 396, 763-770. doi:10.1038/27376
- Hamann A. and Matthaei S. (1996) Regulation of energy balance by leptin. Experimental and Clinical Endocrinology & Diabetes, 104, 293-300. doi:10.1055/s-0029-1211457
- Rumyantsev S.N. (2006) Obesity: A reckoning both for genetic immunity to infection and xenogamy. Medical Hypothesis, 66, 535-540. doi:10.1016/j.mehy.2005.09.014
- Friedman J.M. (2004) Modern science versus the stigma of obesity. Nature Medicine, 10, 563-569. doi:10.1038/nm0604-563
- Farooqi I.S. (2001) Partial leptin deficiency and human adiposity. Nature, 414, 34-35. doi:10.1038/35102112
- Montague C.T., Farooqi I.S., Whitehead J.P., et al. (1997) Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature, 387, 903-908. doi:10.1038/43185
- Stunkard A.J., Harris J.R., Pedersen N.L., et al. (1990) The body-mass index of twins who have been reared apart. The New England Journal of Medicine, 322, 1483-1487. doi:10.1056/NEJM199005243222102
- Rumyantsev S.N. (1983) Constitutional immunity and its molecular-ecological principles [in Russian]. Nauka, Leningrad.
- Nagashima K., Zabriskie J.B.and Lyons M.J. (1992) Virus-induced obesity in mice: association with a hypothalamic lesion. Journal of Neuropathology & Expemental Neurology, 51, 101-109. doi:10.1097/00005072-199201000-00012
- Boizard M., Le Liepvre X., Lemarchand P., et al. (1998) Obesity-related overexpression of fatty-acid synthase gene in adipose tissue involves sterol regulatory element-binding protein transcription factors. The Journal of Biological Chemistry, 273, 29164-29171. doi:10.1074/jbc.273.44.29164
- Atkinson R.L., Dhurandhar N.V., Allison D.B., et al. (2005) Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. International Journal of Obesity, 29, 281-286. doi:10.1038/sj.ijo.0802830
- Atkinson R.L., Whigham L.D., Kim C., et al. (2001) Evaluation of human adenoviruses as an etiology of obesity in chickens. International Journal of Obesity, 25 (Suppl. 2), S12.
- Carbone K.M., Luftig R.B. and Buckley M.R. (2005) Microbial Triggers of Chronic Human Illness. American Academy of Microbiology, Washington, DC, USA.
- So P.W., Herlihy A.H. and Bell J.D. (2005) Adiposity induced by adenovirus 5 inoculation. International Journal of Obesity (Lond), 29, 603-606. doi:10.1038/sj.ijo.0802917
- Vangipuram S.D., Sheel J., Atkinson R.L., et al. (2004) A human adenovirus enhances preadipocyte differentiation. Obesity Research, 12, 770-777. doi:10.1038/oby.2004.93
- Whigham L.D., Israel B.A. and Atkinson R.L. (2006) Adipogenic potential of multiple human adenoviruses in vivo and in vitro in animals. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 290,R190-R194. doi:10.1152/ajpregu.00479.2005
- Gabbert C., Donohue M., Arnold J., et al. (2010) Adenovirus 36 and obesity in children and adolescents. Pediatrics, 126, 721-726. doi:10.1542/peds.2009-3362
- Rumyantsev S.N. (2007) Reasons of the strength and duration of post-infectious immunity. Journal of Chinese Clinical Medicine, 2, 273-281.
- Rumyantsev S.N. (2008) Hereditary Immunity: Fundamental Principles and Exploitation in Life Study and Health Care. Nova Biomedical Books, New York.
- Lee G., Consiglio E., Habig W., et al. (1978) Structure:function studies of receptor for thyrotropin and tetanus toxin: Lipid modulation of effector binding to the glycoprotein receptor component. Biochemical and Biophysical Research Communications, 83, 313-320. doi:10.1016/0006-291X(78)90433-3
- Altrock B.W., Arthur L.O., Massei R.J., et al. (1981) Common surface receptors on both mouse and rat cells distinguish different classes of mouse mammary tumor viruses. Virology, 109, 257-259.
- Jondal M., Klein G., Oldstone MBA, et al. (1976) Association between complement and Epstein-Barr virus receptors on human lymphoid cells. Scandinavian Journal of Immunology, 5, 401-410. doi:10.1111/j.1365-3083.1976.tb00294.x
- Popovic M., Sarin P.S., Robert-Gurroff M., et al. (1983) Isolation and transmission of human retrovirus (human T-cell leukemia virus). Science, 219, 856-859. doi:10.1126/science.6600519
- Enegren B.J., Burness A.T.H. (1977) Chemical structure of attachment sites for viruses on human erythrocytes. Nature, 268, 536-537. doi:10.1038/268536a0
- Ledley F.D., Mullin B.R., Aloj S.M., et al. (1976) Sequence similarity between cholera toxin and glycoprotein hormones: implications for structure activity relationship and mechanism of action. Biochemical and Biophysical Research Communications, 69, 852-859. doi:10.1016/0006-291X(76)90452-6
- Rumyantsev S.N. (2002) Structural predestination of individual diversity in the course and severity of infectious diseases. The Scientific World Journal, 2, 205-216. doi:10.1100/tsw.2002.94
- Rumyantsev S.N., Gerasimov V.K. (2007) The Origin and Functions of Biodiversity in Infectious and NonInfectious Diseases. Focus on Biodiversity Research, Nova Science Publishers, 199-300.
- Lindback S.M., Gabbert C., Johnson B.L., et al. (2010) Pediatric nonalcoholic fatty liver disease: A comprehensive review. Advances In Pediatrics, 57, 85-140. doi:10.1016/j.yapd.2010.08.006
- Rumyantsev S.N. (2006) The origin of individual differences in the course and severity of diseases. The Scientific World Journal, 6, 1674-1704. doi:10.1100/tsw.2006.278
- Rumyantsev S.N. (2003) The intra-individual diversity in senescence. Biogerontology, 4, 171-178. doi:10.1023/A:1024137418419
- Paxton W.A., Martin S.R., Tse D., et al. (1996) Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected dispite multiple high-risk sexual exposure. Nature Medicine, 2, 412-417. doi:10.1038/nm0496-412
- Robson H.G. and Vas S.I. (1972) Resistance of inbred mice to Salmonella typhimurium. Journal of Infectious Diseases, 126, 378-386. doi:10.1093/infdis/126.4.378
- Rumyantsev S.N. (1998) Constitutional and non-specific immunity to infection. Revue Scientifique et Technique de l’Office International des Epizooties, 17, 26-42.
- Vavilov N.I. (1964) Immunity of plants to infectious diseases (In Russian). Nauka, Moscow-Leningrad.
- Novembre J. and Stephens M. (2008) Interpreting principal component analyses of spatial population genetic variation. Nature Genetics, 40, 646-649. doi:10.1038/ng.139
- Coop G., Pickrell J.K., Novembre J., et al. (2009) The role of geography in human adaptation. PLoS Genetics, 5, 16. doi:10.1371/journal.pgen.1000500
- Allison D.B., Edlen-Nezin L. and Clay-Williams G.. (1997) Obesity among African American women: prevalence, consequences, causes, and developing research. Womans Health, 33, 243-274. doi:10.1016/0042-6822(81)90497-9
- Fernandez J.R., Allison D.B. (2003) Understanding racial differences in obesity and metabolic syndrome traits. Nutrition Reviews, 61, 316-319. doi:10.1301/nr.2003.sept.316-319
- World Health Organization. (2010) Obesity and overweight. http://www.who.int/dietphysicalactivity/publications/facts/obesity/en/.