Open Journal of Pediatrics
Vol.08 No.02(2018), Article ID:83826,7 pages
10.4236/ojped.2018.82011
Cushing Syndrome Revealing an Adrenocortical Carcinoma
Bintou Sanogo1, Senkaye-Lagom Aimée Kissou1,2*, Zakari Nikiema2,3, Makoura Barro1,2, Djingri Lankouandé4, Boubacar Nacro1
1Department of Pediatrics, University Hospital Center Souro Sanou (CHUSS), Bobo-Dioulasso, Burkina Faso
2Higher Institute of Health Sciences, Nazi Boni University (UNB), Bobo-Dioulasso, Burkina Faso
3Medical Imaging Service, Bobo-Dioulasso, Burkina Faso
4Department of Internal Medicine, Bobo-Dioulasso, Burkina Faso

Copyright © 2018 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: March 12, 2018; Accepted: April 15, 2018; Published: April 18, 2018
ABSTRACT
Adrenocortical carcinoma is a malignant tumor of the adrenal gland, very rare in pediatric pathology. Authors presented a pediatric case of adrenocortical carcinoma which showed neurology symptoms at 1st visit and Cushing syndrome secondarily. The patient received a ketoconazole-based treatment. The outcome was fatal before the surgical excision of the tumor, because of delayed diagnosis. Early diagnosis and multidisciplinary management of adrenocortical carcinoma could improve the prognosis in children.
Keywords:
Cushing Syndrome, Adrenocortical Carcinoma, Child

1. Introduction
Adrenocortical carcinoma (AC) is a malignant tumor that develops at the expense of the adrenal cortex. It is a very rare tumor in pediatric pathology, representing less than 0.2% of all pediatric tumors [1] . Its global incidence is 0.5 to 2 cases/million inhabitants [2] [3] [4] .
This malignant tumor is most often secreting. It induces an endocrine syndrome related to an increase in adrenal hormone levels. In non-secreting forms, the clinical presentation can be as poor as the chance of discovering tumor. We report a case of AC revealed by Cushing’s syndrome in a 10 years old girl.
2. Observation
The patient, a 10 years old girl, attending school was admitted to pediatrics for disorders of consciousness and tonico-clonic seizures without fever. Interrogation found a notion of recurrent seizures and excessive weight gain during the three to four months preceding the consultation. Physical examination of the patient at admission noted a light coma and a little facio-truncal obesity.
Initial biological assessment was normal: glucose, creatinine and cytobacteriological examination of cerebrospinal fluid (CSF) normal, negative proteinuria, negative HIV serology.
An encephalic computed tomography (CT) scan (Figure 1) performed with and without injection of iodinated contrast medium showed hypodensity ranges of approximately 20 Hounsfield units related to junctional seat cytotoxic edema with vascular repealing after injection, and without mass detected. There was no abnormality of the pituitary region. A peri-critical electroencephalogram (EEG) could not be performed.
The immediate evolution was marked by the appearance of a fever, the further alteration of the state of consciousness with aphasia, as well as the persistence of seizures.
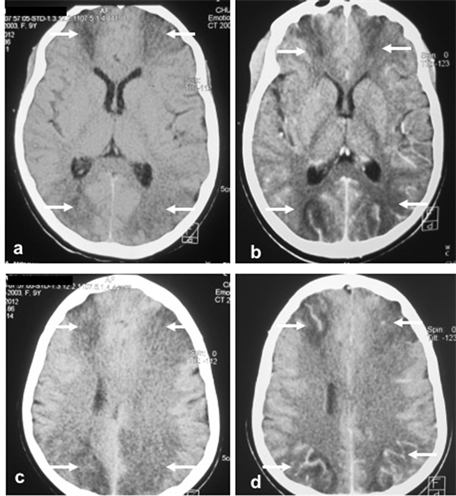
Figure 1. Encephalic CT scan showed hypodensity ranges of approximately 20 Hounsfield units related to junctional seat cytotoxic edema (arrows) with vascular repealing after injection, without mass detected and no abnormality of the pituitary region.
The management was mainly for the safeguarding of major vital functions, as well as the treatment of convulsions, a double broad-spectrum antibiotic therapy and the administration of a cerebral oxygenator. An improvement in the clinical picture was then observed with the recovery of consciousness, the amendment of convulsions and fever in a patient who remained aphasic and bedridden.
The endocrine syndrome became clear about a month later with:
- hypercorticism signs: facio-troncular obesity, skin depigmentation, stretch marks;
- hyperandrogenism signs: clitoral hypertrophy, early pubic hair, acne, hirsutism;
- and the appearance of an abdominal mass at the left hypochondrium.
Some of these signs are observed in Figure 2.
An adrenal assessment was carried out, showing hypercortisolemia and high free 24 hours urinary cortisol. Renal status and blood glucose were within normal limits. All biological tests are summarized in Table 1.
Abdominal ultrasound (Figure 3) revealed a tissue formation developed at the expense of the left adrenal measuring 114 mm × 108 mm × 100 mm, punctuated with calcification in clumps of compatible appearance with AC; kidneys were of normal appearance and there was no live injury.
Abdominal CT-scan (Figure 4) with and without contrast agent injection showed a huge tissue mass adrenal left multilobed evaluated at 1550 cm3, punctuated by different size calcifications, raised after injection, driving the left kidney without invading it and without infiltration of the adjacent fat; there are no
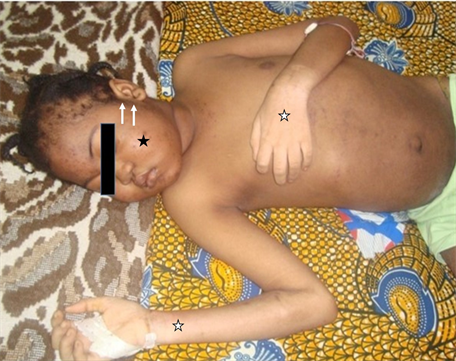
Figure 2. Photograph of the patient showing facio-troncular obesity, skin depigmentation (white stars), stretch marks, acne (black star), hirsutism (arrows).
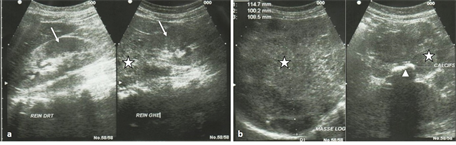
Figure 3. Phtographs of Abdominal ultrasound. A tissue formation developed at the expense of the left adrenal measuring 114 mm × 108 mm × 100 mm (stars in (a) and (b); punctuated with calcification in clumps (head of arrow in (b) of compatible appearance with AC; kidneys were of normal appearance (arrows in (a)) and there was no liver injury.
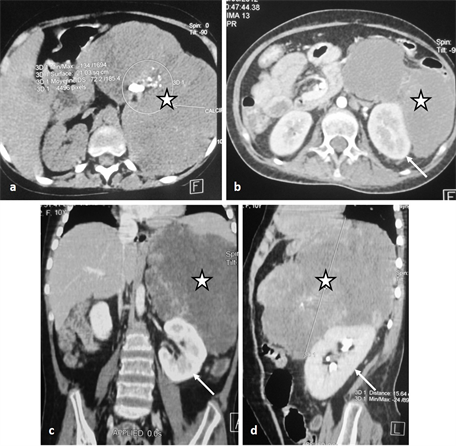
Figure 4. Abdominal CT scan showing a huge tissue mass adrenal left multilobed evaluated at 1550 cm3 (stars in (a)-(d)) punctuated by different size calcifications (circle), raised after injection, driving the left kidney (arrows in (b)-(d)) without invading it and without infiltration of the adjacent fat; there are no deep lymphadenopathies. The aspect was compatible with a left adreno-cortical carcinoma.
deep lymphadenopathies. The aspect was compatible with a left AC. The rest of the exploration was normal.
A treatment based on ketoconazole was undertaken, in the absence of mitotane. Surgical excision of the AC was also indicated and programmed. However, the evolution was marked by the appearance of a malignant hypertension and

Table 1. Synthesis of biological checkup.
the installation of a state of convulsive illness. The death occurred in this chart despite resuscitation measures. Necropsy was not performed because it is not a common practice in our work context.
3. Discussion
Cushing’s syndrome includes all the secondary manifestations to a chronic excess of glucocorticoids which can be associated according to etiologies to a hypersecretion of mineralocorticoids, androgens, or adrenal estrogens.
There are two types: ACTH-dependent Cushing syndrome linked to excessive and inappropriate secretion of ACTH (85% of cases); and ACTH-independent Cushing syndrome, where adrenal secretion is autonomous (15% of cases).
ACTH-independent Cushing syndrome may be due to a benign adrenal (adrenocortical adenoma), or malignant tumor, or a primitive bilateral adrenal (primary pigmented microadenomatosis or macroadenomatous hyperplasia).
Adrenocortical tumors are rare in pediatric pathology. AC is still rare in children [1] [5] , representing less than 0.2% of all pediatric tumors [1] . The peak incidence is around 10 years in children. Female predominance is noted [1] [3] [4] [6] , but would not be the rule when it comes to infants and small children [7] .
In children, the majority of AC are secretory (functional), manifested by virilization, precocious puberty, signs of hypercortisolism; signs of hyper aldosteronism are extremely rare [1] . As for non-secreting corticosteroids, they are often clinically silent and are then fortuitous discovery.
The diagnostic approach during a Cushing’s syndrome is often faced with technical limitations in a context of scarce resources. In fact, after having demonstrated hypercortisolism, the second step is to confirm whether it is ACTH dependent or not [4] . This process was not complete for our patient because the dosage of ACTH and dexamethasone braking tests were not realizable.
On radiology, ultrasonography is the first-line examination for adrenal tumors. However, visualization of a small tumor is not always possible. The existence of intra-tumoral hemorrhage or the presence of necrosis gives a heterogeneous echographic appearance in favor of the malignancy, without being a specific sign [4] . CT is the gold standard examination in the exploration of an adrenal mass [4] . It makes possible to visualize tumors of small diameter of the order of 0.5 to one cm, to look for an attack on neighboring organs (venous tumor extension, hepatic and pulmonary metastases, contralateral surrenal lesions, peri-cortocaval adenopathies) and finally to study the appearance and function of the kidneys. Some signs are in favor of the CS to the scanner: a voluminous mass, a heterogeneous density before and after injection of the contrast medium, and intratumoral calcifications [3] . Epidemiological data tends to show that CS is most often unilateral, and the left adrenal is the most affected [7] .
In our case, the child presented neurological disorders: recurrent convulsions, coma, aphasia, troubles of walking. These events evoked in one hand, brain edema associated with fluid retention related to the hyperaldosteronism; on the other hand, a secondary cerebral localization of the CS although the brain metastases are very rare in this disease [8] [9] . CSF was sterile and chemically normal. Brain CT performed without and with injection of iodinated contrast material showed hypodensity ranges of about 20 Hounsfield units related to a junctional seat cytotoxic edema (Figure 1) with vascular repealing after injection, and without mass detected. A cerebral MRI, and a possible neurosurgical intervention, with histology of a biopsy specimen would have been of great contribution to the diagnosis.
Treatment of none metastatic CS is primarily surgical and consists of adrenalectomy [3] [4] [5] [6]. Although some authors claim that surgery alone is sufficient for well-encapsulated tumors and easy excision, it needs to be supplemented by medical treatment with mitotane, which is a potent synthetic anticortisol and antimitotic [3] [4] . Mitotane is also the adjuvant treatment of choice in cases of metastasis and recurrence [3] [4] . Radiation therapy has a controversial role in AC: some authors recommend it as a palliative treatment for bone metastases while others indicate it in the prevention of local recurrences [3] [4] . Chemotherapy has not proven effective [10] .
Delay in diagnosis, age at the time of diagnosis, signs of endocrine dysfunction, volume and weight of the tumor are all factors of poor prognosis that may explain the fatal outcome of our patient [4] .
4. Conclusion
AC is a rare but redoubtable tumor in children. The diagnosis should be considered in presence of virilization and early signs of puberty. The delay in the diagnosis and the insufficiency of the therapeutic means darken the prognosis in our context.
Cite this paper
Sanogo, B., Aimée Kissou, S.-L., Nikiema, Z., Barro, M., Lankouandé, D. and Nacro, B. (2018) Cushing Syndrome Revealing an Adrenocortical Carcinoma. Open Journal of Pediatrics, 8, 87-93. https://doi.org/10.4236/ojped.2018.82011
References
- 1. Ghazizadeh, F., Ebadi, M., Alavi, S., Arzanian, M., Shamsian, B. and Jadali, F. (2013) Adrenocortical Carcinoma Presenting with Heterosexual Pseudoprecocious Puberty Shortly after Birth: Case Report and Review. Ecancermedicalscience, 7, 289.
- 2. Loncar, Z., Djukic, V., Zivaljevic, V., Pekmezovic, T., Diklic, A., Tatic, S., Dundjerovic, D., Olujic, B., Slijepcevic, N. and Paunovic, I. (2015) Survival and Prognostic Factors for Adrenocortical Carcinoma: A Single Institution Experience. BMC Urology, 15, 43. https://doi.org/10.1186/s12894-015-0038-1
- 3. Dworakowska, D., Drabarek, A., Wenzel, I., Babińska, A., Swiatkowska-Stodulska, R. and Sworczak, K. (2014) Adrenocortical Cancer (ACC)—Literature Overview and Own Experience. Endokrynologia Polska, 65, 492-502.
- 4. Libe, R. (2015) Adrenocortical Carcinoma (ACC): Diagnosis, Prognosis, and Treatment. Frontiers in Cell and Developmental Biology, 3, 45.
- 5. Manual, F.M., Bréaud, S.J., Mas, J.C., Sirvent, N. and Chevallier, A. (2009) Corticosurenaloma in Children. About 2 Cases. Archives of Pediatrics & Adolescent Medicine, 15, 910.
- 6. Hanna, A.M., Tuan, H.P., Askegard-Giesmann, J.R., Grams, J.M., Iqbal, C.W., Stavlo, P. and Moir, C.R. (2008) Outcome of Adrenocortical Tumors in Children. Journal of Pediatric Surgery, 43, 843-849. https://doi.org/10.1016/j.jpedsurg.2007.12.022
- 7. Stratakis, C.A. (2008) Cushing Syndrome Caused by Adrenocortical Tumors and Hyperplasias (Corticotrophin-Independent Cushing Syndrome). Endocrine Development, 13, 117-132. https://doi.org/10.1159/000134829
- 8. Burotto, M., Tageja, N., Rosenberg, A., Mahalingam, S., Quezado, M., Velarde, M., Edgerly, M. and Fojo, T. (2015) Brain Metastasis in Patients with Adrenocortical Carcinoma: A Clinical Series. The Journal of Clinical Endocrinology & Metabolism, 100, 331-336. https://doi.org/10.1210/jc.2014-2650
- 9. Wagner, A.S., Fleitz, J.M. and Kleinschmidt-DeMasters, B.K. (2005) Pediatric Adrenal Cortical Carcinoma: Brain Metastases and Relationship to NF-1, Case Reports and Review of the Literature. Journal of Neuro-Oncology, 75, 127-133. https://doi.org/10.1007/s11060-005-0376-z
- 10. Sipayya, V., Yadav, Y.K., Arora, R., Sharma, U. and Gupta, K. (2012) Virilizing Adrenocortical Carcinoma in a Child: A Rare Enigma. Indian Journal of Endocrinology and Metabolism, 16, 621-623. https://doi.org/10.4103/2230-8210.98025