Open Journal of Internal Medicine
Vol.05 No.03(2015), Article ID:58913,7 pages
10.4236/ojim.2015.53005
Adult Langerhans Cell Histiocytosis: A Rare Etiology of Spinal Cord Compression
Fadila Kouhen1*, Naoual Benhmiddou1, Mohammed Afif1, Fadoua Rais1, Youssef Mahdi2, Basma Khannoussi2, Khadija Bellahamou3, Ibrahim Ghissassi3, Khouloud Boussouni4, Hanan Elkacemi1, Sanaa Elmajjaoui1, Tayeb Kebdani1, Noureddine Benjaafar1
1Department of Radiotherapy, National Institute of Oncology, Rabat, Morocco
2Department of Pathology, National Institute of Oncology, Rabat, Morocco
3Department of Medical Oncology, National Institute of Oncology, Rabat, Morocco
4Department of Radiology, National Institute of Oncology, Rabat, Morocco
Email: *fadila10m@hotmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 20 June 2015; accepted 17 August 2015; published 20 August 2015
ABSTRACT
Langerhans cell histiocytosis is a rare disease involving clonal proliferation of langerhans cells seen in children and young adults. Clinical presentation is variable, ranging from a single location in the bone to severe multivisceral involvement. Moreover, spinal involvement causing myelopathy is even rare and unusual. We report a rare case of adult Langerhans cell histiocytosis in the dorsal spine causing a spinal cord compression associated with a pulmonary process treated by surgery, radiotherapy and systemic therapy with good evolution.
Keywords:
Langerhans Cell Histiocytosis, Adults, Spinal Cord Compression

1. Introduction
Langerhans cell histiocytosis (LCH) is a rare group of idiopathic disorders encompassing large variants of pathologic entities due to diversity of clinical course, evolutionary aspects and prognosis [1] . They all share histologically a significant infiltration of affected tissues by langerhans cells.
Although the features of this disease are well known in children, there are no established guidelines for diagnosis and treatment of adult LCH [2] . Available data were published in pooled retrospective analysis with small numbers of patients and a short follow-up time.
LCH can affect any organ or system. Adult localized forms are dominated by bone and lung disease. Multisystem disease has a highly variable clinical presentation and a poor prognosis [3] .
Different therapeutic approaches can be considered depending on the affected organ, including surgery, radiotherapy and chemotherapy. We report a rare case of adult LCH in the dorsal spine causing a spinal cord compression treated by surgery, radiotherapy and systemic therapy with good evolution.
2. Case Report
A 25-year-old man, non smoker, complained from dorsal pain. He was first treated with analgetics. Because of persistent of the symptoms and the appearance of spastic parapares is of lower limbs. Computed tomography (CT) scan and Magnetic resonance imaging (MRI) were performed.
MRI of the spine showed a spinal cord compression due to bone and epidural tumoral lesions of the posterior wall of the D1 to D5 thoracic vertebras (Figure 1).
A surgical biopsy showed an infiltrate of eosinophilic cells with oval, grooved and convoluted nucleus, associated with eosinophils, lymphocytes and plasma cells (Figure 2).
In immunohistochemical study, these eosinophilic cells expressed CD1a and S100 protein.
Final diagnosis was therefore Langerhans cell histiocytosis of the dorsal spine complicated by spinal cord compression.
A chest CT scan revealed a tumoral process measuring 25 mm at the lower lobe of the left lung evoking a Langerhans cell histiocytosis (Figure 3).
X-rays of the rest of the skeleton, Bone scan and abdominal ultrasound were normal.
The therapeutic decision was an autologeous bone transplantation with a ventral plate osteosynthesis and a decompressive irradiation to the dorsal spine (10 × 3 GY) followed by glucocorticoid and vinblastine based chemotherapy: 10 mg/weeks for 6 weeks.
The evaluation after six courses showed a regression of the process in the pulmonary lower left lobe measuring 16 mm vs. 25 mm and the resolution of pain and paraparesis. The patient was regularly followed with clinical and imaging examinations for 26 months. He had no subjective complaints, symptoms of recurrence or worsening and no appearance of another lesion (Figure 4 and Figure 5).
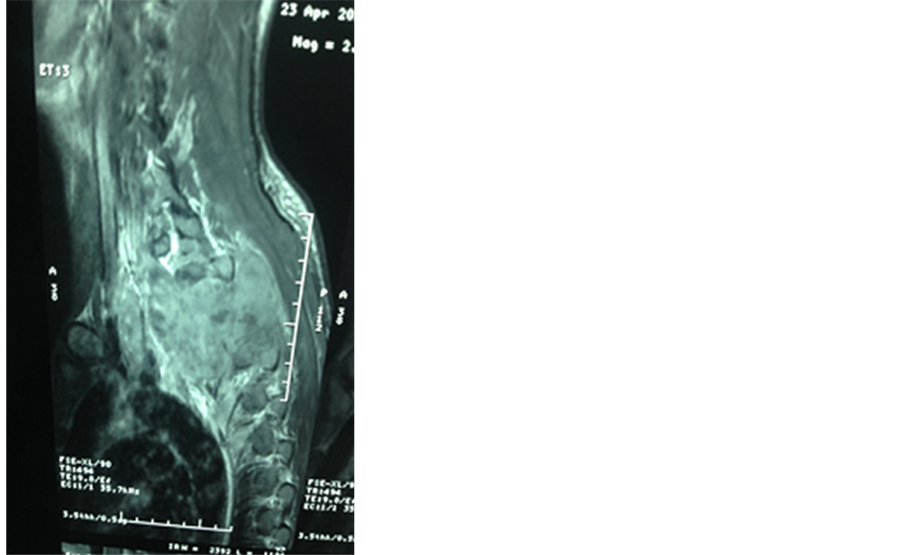
Figure 1. T1 and T2 magnetic resonance imaging (MRI) images show cord compressing tumoral mass at D1 to D5 thoracic vertebras.

Figure 2. Microscopic aspect. It consists of Langerhans cells with oval, grooved and convoluted nucleus and slightly eosinophilic cytoplasm, associated with eosinophils, lymphocytes and plasma cells (H & E × 400).
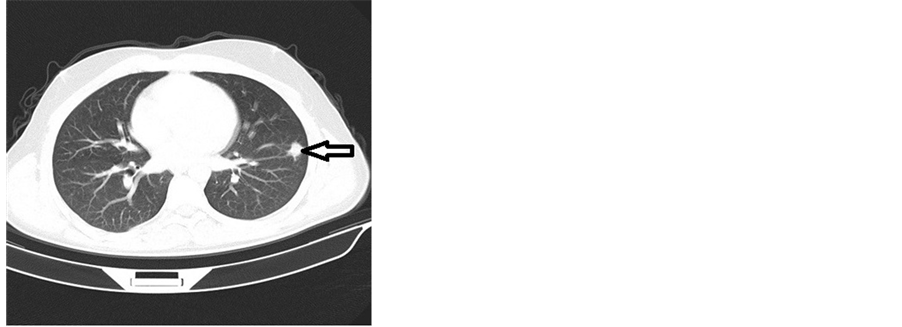
Figure 3. Computed tomography (CT) scan of the chest showing tumoral processes at the lower lobe of the left lung.

Figure 4. Computed tomography (CT) scan of the spine after the treatment.

Figure 5. Computed tomography (CT) scan of the chest after the treatment.
3. Discussion
Langerhans cell histiocytosis (LCH) is an orphan disease. In 1868, Langerhans discovered the epidermal dendritic cells that now bear his name [4] .
It affects mainly children with a peack incidence from 1 to 5 years. The incidence is estimated to be 1/20,000 per year with a male predilection (male-to-female ratio of 2:1) [5] .
Adult LCH is rarer and its reported incidence is around one to two cases per million people per year [6] .
The etiology of Langerhans cell histiocytosis (LCH) remains unknown. It’s may be induced by a viral infection (Specifically, human herpesvirus 6 (HHV-6), a defect in intercellular communication (T cell-macrophage interaction), and/or a cytokine-driven process mediated by tumor necrosis factor, IL-11, and leukemia inhibitory factor [7] - [9] .
The working group of the Histiocyte Society has divided histocytic disorders into 3 groups: dendritic cell histiocytosis, macrophage-related disorders, and malignant histiocytosis [10] . LCH belongs in group 1 and encompasses a number of diseases.
Many tissues are subject of a polymorph inflammatory infiltrate of atypical histiocyts, lymphocytes, and macrophages mixed with eosinophils and neutrophils with a granulomatous disposal [11] . The accumulation of these cells causes the classic clinical and imaging symptoms such as lytic bone lesions, skin rashes, lymphadenopathy, splenomegaly, and organ dysfunction of the pituitary gland, respiratory function, liver, and hematologic system [12] .
Any organ can be affected, but lesions mainly affect the tissues where, in the normal state, there are Langerhans cells or their precursors. Among the most frequently involved organs there is bone, skin, the hypothalamic-pituitary axis and lungs [13] [14] .
The bone lesion can be unique or multiple, they are located mainly in the flat bones most commonly in the skull (51%), the jaw (30%) causing loss of teeth. But can less frequently affect tubular bones (17%), pelvis (13%), and ribs (6%) [13] . The vertebral involvement is seen in 7% of cases, and the dorsal vertebras are the most commonly affected [15] . The development of spinal cord compression is unusual.
Named initially histiocytosis X, it included three clinical subtypes: eosinophilic granuloma, which comprises only bone or lung involvement and has a favorable outcome without treatment or with a local treatment; Hand-Schûller-Christian’s disease, affects children more than two to three years, involves multiple organs and typically combines diabetes insipidus, cranial bone gaps and exophthalmos; and Letterer-Siwe’s disease that affects children under 2 years old, which typically involves the abdominal viscera, has the most severe manifestations and remains of a poor prognosis [16] [17] .
Currently, the term Langerhans cell histiocytosis is used, because the histiogenesis basis has been clarified. Recently, the Writing Group of the Histiocyte Society stratified the disease according to the number and type of organs involved, defining single system disease and multisystem disease.
Further, we distinguish multisystem disease low risk patients (with liver, spleen and bone marrow involvement) and high-risk patients (with skin, bone, lung, lymph nodes, gastrointestinal tract, pituitary gland, central nervous system involvement) [18] . The reported case is considered high risk patients with multisystem disease (bone and lung).
Clinical manifestations of LCH depend on the organ affected. Bone lesions are usually asymptomatic, but bone pain and a soft tissue mass may occur. The presented patient had typical signs of the spinal cord compression: pain and neurological deficit [15] .
Clinical presentation in isolated pulmonary LCH is polymorphic. Asymptomatic disease is discovered on routine chest radiography in 25% of cases as has been the case in our patient.
Nearly two-thirds of patients have functional signs usually dominated by non-productive cough. The presence of an inaugural chest pain is rare and should lead to investigate a pneumothorax revealing the affection in 10% to 20% of cases especially in young men [19] [20] .
Imaging is based predominantly on radiography in the diagnostic and staging of LCH [21] .
Radiological diagnostic examination should at least include entire skeletal X ray, a chest X ray, and an abdominal ultrasound. Further imaging tools are necessary according to clinical presentation [13] .
In bone LCH, entire skeletal X ray is the main investigation. It shows typically single or multiple osteolytic lesions round or oval-shaped with well-defined contours [21] , sometimes associated with peripheral condensation. Bone involvement can be accompanied by a cortical blowing and a periosteal reaction or endosteal scalloping, responsible for the “budding appearance” on Computed tomography (CT) or magnetic resonance imaging (MRI) and evoking a differential diagnosis with Ewing’s sarcoma, a tuberculoma, or osteomyelitis [22] . Ver- tebral lesions imaging shows typically a vertebral destructing lesion respecting the posterior wall and intervertebral discs, responsible of characteristic vertebra plana appearance [21] [23] .
CT and MRI used for better analysis of cortical erosion and tissue involvement. In presence of neurological symptoms, MRI is indicated to investigate spinal cord and epidural involvement [24] [25] as has been the case in our patient.
T99 scintigraphy is not specific but sometimes complementary. The contribution of positon emission tomography is poorly defined [21] .
In pulmonary LCH, computed tomography is the diagnostic imaging procedure of choice.
In the reported case, diagnosis is evocated in presence of nodes. Typically ring figures are found in early stages. Pulmonary fibrosis that generally respects inferior lobs is usually restricted to advanced forms [26] .
The diagnosis is based on histologic and immunophenotypic examination of a lesional biopsy showing a granuloma Langerhans cells whose appearance differs according to the evolutionary stage of the disease and the envolved tissue. On optic microscope, Langerhans cells are recognized by their convoluted core, blade and slightly eosinophilic cytoplasm, containing little or no phagocytic particles [27] . An infitration by lymphocytes and eosinophilic granulocyts forming pseudoabscesses is characteristic. Definitive diagnosis can only be made with immunochemical study showing that cells stain positively with CD1a or CD 207 [28] . Staining for S-100 protein is non specific for Langerhans cells. Birbeck granules in electron microscopy are also possible, very specific, but time consuming and expensive.
No consensus exists for the optimal therapy for Langerhans cell histiocytosis, particularly in the case of multisystem organ disease. Generally, the choice of therapeutic regimen varies according to involved organs. Single system disease has an excellent outcome, usually without need for systemic therapy [13] . Disease can resolve after simple waiting or local treatment. Smoking cessation is an important intervention in cases of pulmonary LCH [29] . A bony lesion requires generally no treatment and may heal spontaneously or after curettage. Pain responds to local injection of methylprednisolone acetate [28] . Operative hemicorporectomy or semitotal corporectomy is reserved to cases with neurological symptoms or deficits. Due to medullary compression, therapeutic decision for our patient an autologeous bone transplantation with a ventral plate osteosynthesis.
Indications of radiotherapy are not yet well codified. Some authors recommend that radiation therapy should be restricted to emergency situations such as optic nerve or spinal cord compression because of the risk of secondary cancer [30] [31] and others emphasize the effectiveness of radiation therapy alone or in combination with corticosteroids in uni- or multifocal osseous single-system disease with good results in most series [32] [33] .
The dose recommendation for radiotherapy is still controversial. There is a wide dose range from 1, 4 Gy up to 45 Gy, but usually the dose varies between 10 to 20 Gy is recommended in adults. In the reported case, the indication of radiotherapy was spinal cord compression with dose of 30 Gy in 10 fractions. The local control rates ranged from 75% - 100%, complete remission from 79% - 100%, respectively [34] .
Patients showing progressive disease receive corticotherapy. Even if relapse occurs often in adult LCH patients, chemotherapy is reserved as salvage treatment for progressive disease after observation, smoking cessation and prednisone based therapy.
In contrast, forms with disseminated disease have a poor prognosis and require systemic treatment. They should receive a prednisone and vinblastine based therapy during 6 weeks, and then continue therapy with 6 mercaptopurine, prednisone, and vinblastine for 6 to 12 months. In the case described here, he received glucocorticoid and vinblastine based chemotherapy: 10 mg/weeks for 6 weeks.
The prognosis depend mainly on patient, disease, and therapy related factors. Single sytem disease patients have a better outcome than patients with multisystem disease. The first group enjoys a 5-year event free survival of 100% while it approaches 91.7% for patients with multisystem disease.
Survival did not differ significantly among patients with multisystem disease, with or without liver or lung involvement) 5-year survival 93.6% (95% CI 84.7 - 97.4) versus 87.5% (95% CI 65.5 - 95.9), respectively; P value 0.1) [27] .
4. Conclusion
Due to low incidence of LCH in children and adults, patients must be enrolled on multi-national clinical trials, whenever possible, to resolve some of the outstanding issues and improve our knowledge of the optimal therapeutic strategies and long-term outcomes.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgements
All the authors are thankful for providing the necessary facilities for the preparation the manuscript.
Special thanks are due to the Faculty of Medicine and Pharmacy of Rabat.
Cite this paper
FadilaKouhen,NaoualBenhmiddou,MohammedAfif,FadouaRais,YoussefMahdi,BasmaKhannoussi,KhadijaBellahamou,IbrahimGhissassi,KhouloudBoussouni,HananElkacemi,SanaaElmajjaoui,TayebKebdani,NoureddineBenjaafar, (2015) Adult Langerhans Cell Histiocytosis: A Rare Etiology of Spinal Cord Compression. Open Journal of Internal Medicine,05,21-28. doi: 10.4236/ojim.2015.53005
References
- 1. Khung, S., Budzik, J.F., Amzallag-Bellenger, E., Lambilliote, A., Soto Ares, G., Cotten, A., et al. (2013) Skeletal Involvement in Langerhans Cell Histiocytosis. Insights Imaging, 4, 569-579.
http://dx.doi.org/10.1007/s13244-013-0271-7 - 2. Girschikofsky, M., Arico, M., Castillo, D., Chu, A., Doberauer, C., Fichter, J., et al. (2013) Management of Adult Patients with Langerhans Cell Histiocytosis: Recommendations from an Expert Panel on Behalf of Euro-Histio-Net. Orphanet Journal of Rare Diseases, 8, 72.
http://dx.doi.org/10.1186/1750-1172-8-72 - 3. Broadbent, V., Egeler, R.M. and Nesbit Jr., M.E. (1994) Langerhans Cell Histiocytosis: Clinical and Epidemiological Aspects. Br J Cancer Suppl, 23, S11-S6.
- 4. Langerhans, P. (1868) Uber die nerven der menschlichen haut. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin, 44, 325-327.
http://dx.doi.org/10.1007/BF01959006 - 5. Robitaille, N. and Hume, H.A. (2006) Blood Components and Fractionated Plasma Products: Preparations, Indications, and Administration. Pediatric Hematology, 3, 693-708.
http://dx.doi.org/10.1002/9780470987001.ch32 - 6. Andersson By, U., Tani, E., Andersson, U. and Henter, J.I. (2004) Tumor Necrosis Factor, Interleukin 11, and Leukemia Inhibitory Factor Produced by Langerhans Cells in Langerhans Cell Histiocytosis. Journal of Pediatric Hematology/Oncology, 26, 706-711.
http://dx.doi.org/10.1097/00043426-200411000-00004 - 7. Kawakubo, Y., Kishimoto, H., Sato, Y., Yanagimoto, K., Tsuruta, T., Ogawa, Y., et al. (1999) Human Cytomegalovirus Infection in Foci of Langerhans Cell Histiocytosis. Virchows Archiv, 434, 109-115.
http://dx.doi.org/10.1007/s004280050313 - 8. Csire, M., Mikala, G., Jako, J., Masszi, T., Janosi, J. and Dolgos, J. (2007) Persistent Long-Term Human Herpesvirus 6 (HHV-6) Infection in a Patient with Langerhans Cell Histiocytosis. Pathology and Oncology Research, 13, 157-160.
http://dx.doi.org/10.1007/BF02893493 - 9. Satter, E.K. and High, W.A. (2008) Langerhans Cell Histiocytosis: A Review of the Current Recommendations of the Histiocyte Society. Pediatric Dermatology, 25, 291-295.
http://dx.doi.org/10.1111/j.1525-1470.2008.00669.x - 10. Beverley, P.C., Egeler, R.M., Arceci, R.J. and Pritchard, J. (2005) The Nikolas Symposia and Histiocytosis. Nature Reviews Cancer, 5, 488-494.
http://dx.doi.org/10.1038/nrc1632 - 11. Allen, C.E., Li, L., Peters, T.L., Leung, H.C., Yu, A., Man, T.K., et al. (2010) Cell-Specific Gene Expression in Lesions Has a Different Profile Compared with Normal Epidermal Langerhans Cells. The Journal of Immunology, 184, 4557-4567.
http://dx.doi.org/10.4049/jimmunol.0902336 - 12. McClain, K.L. and Allen, C.E. (2010) Inflammatory and Malignant Histiocytosis. McGraw Hill Medical, New York, 1047-1064.
- 13. Ghorbel, I.B., Boukhris, I., Kchir, M.N., Abderrahmane, K., Khanfir, M., Lamloum, M., et al. (2008) Langerhans’ Cell Histiocytosis of the Dorsal Spine. A Rare Etiology of Spinal Cord Compression. La Revue de Médecine Interne, 29, 58-62.
http://dx.doi.org/10.1016/j.revmed.2007.08.019 - 14. Baillet, A., Grange, L., Lafaix, P.A., Gaudin, P. and Juvin, R. (2007) Histiocytose de Langerhans et radiculalgie. Revue Du Rhumatisme, 74, 289-293.
http://dx.doi.org/10.1016/j.rhum.2006.05.026 - 15. Simanski, C., Bouillon, B., Brockmann, M. and Tiling, T. (2004) The Langerhans’ cell Histiocytosis (Eosinophilic Granuloma) of the Cervical Spine: A Rare Diagnosis of Cervical Pain. Magnetic Resonance Imaging, 22,589-294.
http://dx.doi.org/10.1016/j.mri.2004.01.006 - 16. Lichtenstein, L. (1953) Histiocytosis X: Integration of Eosinophilic Granuloma of Bone, Letterer-Siwe Disease, and Schuller-Christian Disease as Related Manifestations of a Single Nosologic Entity. A.M.A. Archives of Surgery, 56, 84- 102.
- 17. Hoover, K.B., Rosenthal, D.I. and Mankin, H. (2007) Langerhans Cell Histiocytosis. Skeletal Radiology, 36, 95-104.
http://dx.doi.org/10.1007/s00256-006-0193-2 - 18. Numata, T., Okubo, Y., Uchiyama, M., Takeuchi, Y., Muro, M., et al. (2015) Adult Case of Langerhans Cell Histiocytosis. The Journal of Dermatology, 42,107-108.
http://dx.doi.org/10.1111/1346-8138.12732 - 19. Tazi, A. (2006) Adult Pulmonary Langerhans’ Cell Histiocytosis. European Respiratory Journal, 27, 1272-1285.
http://dx.doi.org/10.1183/09031936.06.00024004 - 20. Vassallo, R., Ryu, J.H., Schroeder, D.R., Decker, P.A. and Limper, A.H. (2002) Clinical Outcomes of Pulmonary Lan- gerhans’-Cell Histiocytosis in Adults. The New England Journal of Medicine, 346, 484-490.
http://dx.doi.org/10.1056/NEJMoa012087 - 21. Khung, S., Budzik, J.F., Amzallag-Bellenger, E., et al. (2013) Skeletal Involvement in Langerhans Cell Histiocytosis. Insights into Imaging, 4, 569-579.
http://dx.doi.org/10.1007/s13244-013-0271-7 - 22. Vadivelu, S., Mangano, F.T., Miller, C.R. and Leonard, J.R. (2007) Multifocal Langerhans Cell Histiocytosis of the Pediatric Spine: A Case Report and Literature Review. Childs Nervous System, 23,127-131.
http://dx.doi.org/10.1007/s00381-006-0172-x - 23. Azouz, E.M., Saigal, G., Rodriguez, M.M. and Podda, A. (2005) Langerhans’ Cell Histiocytosis: Pathology, Imaging and Treatment of Skeletal Involvement. Pediatric Radiology, 35, 103-115.
http://dx.doi.org/10.1007/s00247-004-1262-0 - 24. Sutton, D. (2011) Textbook of Radiology and Imaging. Churchill Livingstone, London, Vol. 7, 1340-1344.
- 25. Yochum and Rowe’s (2005) Essential of Skeletal Radiology. Lippincott/Williams & Wilkins, Philadelphia, Vol. 3, 1533-1534.
- 26. Guillerman, R.P. (2010) Imaging of Childhood Interstitial Lung Disease. Pediatric Allergy, Immunology and Pulmonology, 23, 43-68.
http://dx.doi.org/10.1089/ped.2010.0010 - 27. Geissmann, F., Emile, J.F., Donadieu, J., Andry, P., Thomas, C. and Brousse, N. (1997) Aspects cliniques et physiopathologiques de l’histiocytose langerhansienne. Une proliferation clonale de cellules dendritiques de Langerhans. Hématologie, 3, 33-43.
- 28. Windebank, K. and Nanduri, V. (2009) Langerhans Cell Histiocytosis. Archives of Disease in Childhood, 94, 904-908.
http://dx.doi.org/10.1136/adc.2007.125872 - 29. Lin, M.W., Chang, Y.L., Lee, Y.C., Cheng, H.L., Chen, J.S. and Hsu, H.H. (2009) Pulmonary Langerhans Cell Histiocytosis. Lung, 187, 261-262.
http://dx.doi.org/10.1007/s00408-009-9151-9 - 30. Thomas, C. and Donadieu, J. (2008) Histiocytose langerhansienne: Prise en charge thérapeutique. Archives de Pédiatrie, 15, 529-531.
http://dx.doi.org/10.1016/S0929-693X(08)71822-0 - 31. Sahin, F., Ozen, K.P., Vural1, F., Kamer, S., Argin, M., Anacak, Y., et al. (2007) Retrospective Analysis of Seven Patients with Adult-Onset Langerhans Cell Histiocytosis Syndromes: A Single Center experience. Turkish Journal of He- matology, 24, 75-79.
- 32. Cassady, J.R. (1987) Current Role of Radiation Therapy in the Management of Histiocytosis-X. Hematology/Oncology Clinics of North America, 1, 123-129.
- 33. Brady, L.W. (2008) Langerhans Cell Histiocytosis. Springer Verlag, Berlin, 397-423.
- 34. Micke, O. and Seegenschmiedt, M.H. (2002) Consensus Guidelines for Radiation Therapy of Benign Diseases: A Multicenter Approach in Germany. International Journal of Radiation Oncology Biology Physics, 52, 496-513.
http://dx.doi.org/10.1016/S0360-3016(01)01814-4
NOTES

*Corresponding author.