Open Journal of Internal Medicine
Vol.2 No.4(2012), Article ID:25457,4 pages DOI:10.4236/ojim.2012.24030
Unusual cause of syncopal episodes
![]()
1Department of Internal Medicine “B” Ziv Medical Center, Safed, Israel
2Faculty of Medicine in the Galilee, Bar-Ilan University, Safed, Israel
3Obstetrics and Gynecology, Western Galilee Hospital, Nahariya, Israel
4Internal Medicine “A” Department, Western Galilee Hospital, Nahariya, Israel
Email: *raymond.f@ziv.health.gov.il
Received 29 June 2012; revised 9 October 2012; accepted 18 November 2012
Keywords: Syncope; Hypotension; Flushing; Mastocytosis
ABSTRACT
We report a 43-year-old man who had been experiencing recurrent attacks of syncope for several years, and a search for the causes behind these lapses in consciousness was fruitless for many years. The patient was admitted to our department of internal medicine because of the symptoms flushing, hypotension and episodes of syncope. After a careful anamnesis and then a comprehensive investigation, we reached the definitive diagnosis of the infrequent disease “systemic mastocytosis”.
1. INTRODUCTION
Episodic flushing with hypotension is common in a variety of relatively uncommon endocrine disorders like carcinoid syndrome, medullary carcinoma of thyroid and systemic mastocytosis. Some patients with pheochromocytoma may have flushing accompanied by paroxysmal palpitations and hypertension. These diseases can cause recurrent syncope and flushing due to release of histamine and vasoactive prostaglandins (PGD2) [1-3]. Systemic mastocytosis is an uncommon disease; its incidence and prevalence are unknown. It is characterized by an increased number of tissue mast cells and mostly is limited to cutaneous manifestations [4]. The correct diagnosis could be missed, especially because of the spontaneous disappearance of symptoms after a short time, leaving no obvious need for further investigation. The patient described here was admitted to our department of internal medicine because of the flushing and hypotension that was documented by an ambulance paramedic.
2. CASE REPORT
A 43-year-old man, heavy smoker, was admitted to the department of internal medicine for investigation because of his long history of recurrent episodes of syncope. Each episode was expressed by hypotension, palpitations, sensation of pressure in his chest and flushing. These episodes repeated, approximately every three months. Due to the working conditions created by his job (sailor), he had been examined in various emergency rooms in different countries. He had a history of chronic gastritis diagnosed by upper gastrointestinal radiography with barium followed by gastroduodenoscopy and gastric biopsy during his first admission into the medical department 6 years ago. Since that time he has been treated with ranitidine®. At that time comprehensive investigation for carcinoid syndrome, thyroid medullary carcinoma and pheochromocytoma was negative. Additionally, cardiac and neurologic investigations were negative and as far as we understood from the patients’ history and past work-ups, systemic mastocytosis was not a differential diagnosis. There was no history of fever, weight loss, sweats, diarrhea, dyspnea, wheezing, bone aches, cardiac murmur, cutaneous abnormalities, pruritus, or allergic reactions.
On first admission, the patient was alert, oriented, his body temperature was 36.6˚C, pulse was regular at 100/m, and respiratory rate was 14. Blood pressure was initially 80/60 mmHg and rose to 113/65 mmHg, after a subcutaneous injection of epinephrine that was given by the ambulance personnel on his way to the emergency room in our hospital. On arrival, a slight flushing of the face was still-apparent; otherwise, the physical examination was normal. Recurrent episodes and admissions were of the same nature. On his last admission prior to six months, the patient had the same complaints and symptomatic treatment was prescribed. Laboratory data showed:
Hemoglobin was 13.4 g/dl; the white cell count was 9.9 × 103/l with normal differentiation and normal platelet count. Blood urea nitrogen, creatinine, glucose and liver function tests were all within normal limits. Urine 24-hour collection for 5-hydroxyindolacetic acid (5- HIAA), vanillylmandelic acid (VMA) and total metanephrines were normal. No hypoglycemia was documented during a prolonged glucose tolerance test. Chest X-ray films revealed clear lungs and normal cardiac size. On electrocardiogram, the patient had a sinus tachycardia at a rate of 110, with a mean QRS axis +60. Echo-cardiographic study and multiple Holter monitoring for 24 hours were normal. A stress test showed that the heart rate rose to 180, with a rise in blood pressure to 175/95 mmHg without evidence of ischemia. Carotid sinus hypersensitivity was negative and no sign of orthostatic hypotension present, table tilt test was negative and other investigation for eventual pulmonary embolism was negative.
On the fifth day after admission, and because the patient’s work-up was negative, although extensive, we put a systemic mastocytosis on the differential diagnosis list and we performed a bone marrow examination and found paratrabecular aggregates of mast cells mixed with small lymphocytes. Those mast cells were round to spindleshaped with purple granules in the cytoplasm. The bony trabeculae and the rest of the bone marrow cellular components were normal (Figure 1).
Thoracic and abdominal computerized tomography scans (CT) were normal. Radionuclide 99 m TC-methylene diphosphate bone scan showed a diffuse increased bone activity. On the basis of these findings: clinical presentation of symptoms and signs, bone marrow biopsy and radio nucleid 99 m TC-methylene diphosphate bone scan, the diagnosis of systemic mastocytosis was made. Treatment with lorastine® (Loratadine) (10 mg/day) was started on the 5th day after admission and on the 9th day the patient was discharged with permanent daily dose of 10 mg.
Since the patient’s discharge from the hospital 24 months ago, no further attacks were encountered.
3. DISCUSSION
Systemic mastocytosis is a rare neoplastic disease characterized by abnormal proliferation of mast cells and their CD34+ that may infiltrate various organs including skin, gastrointestinal tract, liver, spleen, bone marrow and lymph nodes. The clinical presentation of systemic mastocytosis disease (SMCD) is that of a wide range according to the organ or organs involved [1]. Urticaria pigmentosa associated with Darier’s sign (wheal and flare after gently stroking a skin lesion) is considered a pathognomonic sign of mastocytosis. In the rare cases of systemic mastocytosis, the patients have recurrent attacks of flushing, hypotention or syncope and palpitations that usually last 15 - 30 minutes. That may be accompanied with headache, dyspnea, nausea and vomiting, parasthesias or pruritus. Some patients may present with bone pain, pathologic fractures, and osteoporosis, or with hema
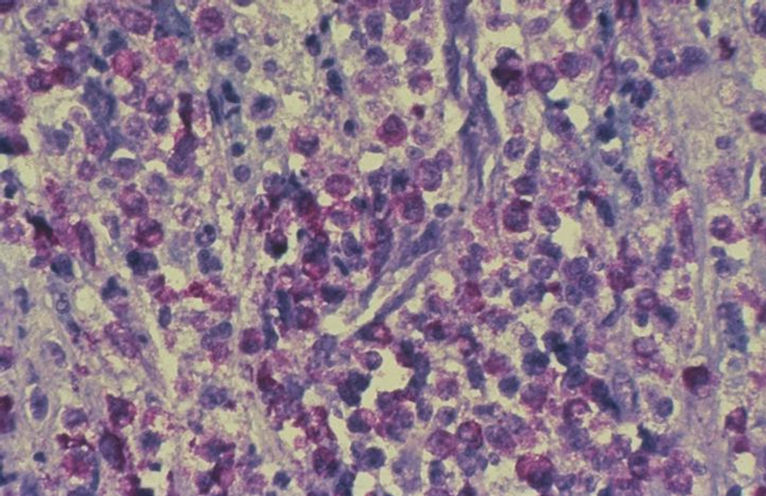
Figure 1. High magnification of a marrow lesion in systemic mastocytosis. The mast cells have a spindle appearance. There are numerous interspersed eosinophils and paratrabecular aggregates of mast cells mixed with small lymphocytes.
tologic or solid malignancy [2,3]. Systemic mastocytosis without urticaria pigmentosa was reported in rare cases. The rarity of the disease and the diversity of its clinical presentation may obscure its inclusion in the differential diagnosis. However, the definitive diagnosis of SMCD is histologic demonstration of proliferative mast cells infiltrating the tissue involved. Biopsy of skin lesion in urticaria pigmentosa is usually diagnostic and in systemic mastocytosis, bone marrow biopsy is essential [1,4]. The major mediators of symptoms in this disease are PGD2 and histamine. PGD2 as a vasodilator is responsible for flushing, hypotension and syncope, while the histamine is the cause of cutaneous and gastrointestinal symptoms: reflux esophagitis and peptic ulcerations. Biochemical evidence of mast cell activation by measuring the serum and urine histamine and prostaglandin D2 and their metabolites only during the attack, may contribute to the diagnosis, but that is not easy because the patients usually arrive at the hospital after the attack. Bone scans and gastrointestinal endoscopies, in some patients, are also indicated [4].
The classification of systemic mastocytosis to malignant and benign disease cannot be predicted according to histologic appearance of mast cell population because the patients may have a change in the course of their disease and may possibly die because of hematologic disorders or solid malignancy. Investigators have found that the distinction between benign and malignant SMCD is not always clear-cut [1]. A WHO consensus classification for mastocytosis exists, which is widely accepted and includes three major categories: 1) Cutaneous mastocytosis (CM), a benign disease in which MC infiltration is confined to the skin, is preferentially seen in young children and exhibits a marked tendency to regress spontaneously; 2) Systemic mastocytosis (SM) which is commonly diagnosed in adults and includes four major subtypes: a) indolent SM (ISM, the most common form involving mainly skin and bone marrow); b) a unique subcategory termed SM with an associated non-mast cell clonal hematological disease (SM-AHNMD); c) aggressive SM usually presenting without skin lesions; and d) MC leukemia, probably representing the rarest variant of human leukemias; 3) The extremely rare localized extracutaneous MC neoplasms, either presenting as malignancy (MC sarcoma) or as benign tumor termed extracutaneous mastocytoma. Diagnostic criteria for mastocytosis are available and are widely accepted. SM criteria include one major criterion (multifocal compact tissue infiltration by MC) and four minor criteria: 1) prominent spindling of MC; 2) atypical immunophenotype of MC with coexpression of CD2 and/or CD25 (antigens which have not been found to be expressed on normal/reactive MC); 3) activating (somatic) point mutations of the c-kit protooncogene usually involving exon 17, with the imatinibresistant type D816V being most frequent; and 4) persistently elevated serum tryptase level (>20 ng/ml). To establish the diagnosis of SM, at least one major and one minor criterion, or at least three minor criteria, have to be fulfilled [5]. Accordingly, the prognosis depends on the course of the disease, so, the range of survival may be less than two years and more than ten years.
The patient presented here has all the criteria for the diagnosis of SMCD: recurrent attacks of flushing, hypotension and syncope during the last fourteen years, gastrointestinal symptoms proved by gastric endoscopy and biopsy, and the histology of bone marrow biopsy.
The treatment in our case was the same as it is recommended and included H1 and H2 blockers. High-dose aspirin is used to block prostaglandin synthesis but because of the chronic gastritis of our patient we did not recommend this kind of treatment. Subcutaneous epinephrine is used to abort an acute vasomotor attack [6].
Since our patient was discharged from the hospital, he has been followed up at our outpatient clinic and reported no further complaints. It should be mentioned that our patient drinks alcohol and that these attacks might well have been triggered by this drinking behavior. In any case of recurrent urticaria or flushing associated with hypotension, a careful anamnesis and then a comprehensive investigation are needed to reach the definitive diagnosis as in our described case.
REFERENCES
- Travis, W.D., Li, C.-Y., Bergstralh, E.J., Yam, L.T. and Swee, R.G. (1988) Systemic mast cell disease: Analysis of 58 cases and literature review. Medicine, 67, 345-365.
- Roberts, L.J., Hubbard, W.C. and Bloomgarden, Z.T. (1979) Prostaglandins: Role in the humoral manifestations of medullary carcinoma of the thyroid and inhibition by somatostatin. Transactions of the Association of American Physicians, 92, 286-291.
- Suchard, J.R. (1997) Recurrent near-syncope with flushing. Academic Emergency Medicine, 4, 718-724. doi:10.1111/j.1553-2712.1997.tb03767.x
- Metcalfe, D.D. (1991) Clinical advances in mastocytosis: An interdisciplinary roundtable discussion. Journal of Investigative Dermatology, 96, 1S-65S. doi:10.1111/1523-1747.ep12468869
- Horny, H.P., Sotlar, K. and Valent, P. (2007) Mastocytosis: State of the art. Pathobiology, 74, 121-132. doi:10.1159/000101711
- Bateman, H.E., Schroff, V., Centeno, L.V. and Bielory, L. (1995) Systemic mastocytosis: A diagnostic challenge. Annals of Allergy, 74, 379-386.
NOTES
*Corresponding author.