Stem Cell Discovery
Vol.05 No.03(2015), Article ID:58449,13 pages
10.4236/scd.2015.53003
Isolation and Characterization of Multipotent and Pluripotent Stem Cells from Human Peripheral Blood
Ciro Gargiulo1*, Van Hung Pham2, Nguyen Thuy Hai1, Kieu C. D. Nguyen3, Pham Van Phuc4, Kenji Abe5, Veronica Flores6, Melvin Shiffman7
1Division of Internal Medicine, The Human Medicine International Clinic, Ho Chi Minh City, Vietnam
2Molecular Diagnostics Department, Nam Khoa-Biotek Laboratory, Ho Chi Minh City, Vietnam
3Division of Pathology, The Human Medicine International Clinic, Ho Chi Minh City, Vietnam
4Department of Stem Cells and Regenerative Medicine, University of Natural Science, Ho Chi Minh City, Vietnam
5Department of Pathology, National Institute of Infectious Diseases, Tokyo, Japan
6Sol Price School of Public Policy, University of Southern California, Los Angeles, USA
7Section of Surgery, Newport Specialty Hospital, Tustin, USA
Email: *dr.ciro@humanmedicine.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 26 April 2015; accepted 27 July 2015; published 30 July 2015
ABSTRACT
Stem cells are commonly classified based on the developmental stage from which they are isolated, although this has been a source of debate amongst stem cell scientists. A common approach classifies stem cells into three different groupings: Embryonic Stem Cells (ESCs), Umbilical Cord Stem Cells (UCBSCs) and Adult Stem Cells (ASCs), which include stem cells from bone marrow (BM), fat tissue (FT), engineered induced pluripotent (IP) and peripheral blood (PB). By definition stem cells are progenitor cells capable of self-renewal and differentiation hypothetically “ab infinitum” into more specialized cells and tissue. The main intent of this study was to determine and characterize the different sub-groups of stem cells present within the human PB-SCs that may represent a valid opportunity in the field of clinical regenerative medicine. Stem cells in the isolated mononucleated cells were characterized using a multidisciplinary approach that was based on morphology, the expression of stem cell markers by flowcytometry and fluorescence analysis, RT-PCR and the capacity to self-renew or proliferate and differentiate into specialized cells. This approach was used to identify the expression of hematopoietic, mesenchymal, embryonic and neural stem cell markers. Both isolated adherent and suspension mononucleated cells were able to maintain their stem cell properties during in-vitro culture by holding their capacity for proliferation and differentiation into osteoblast cells, respectively, when exposed to the appropriate induction medium.
Keywords:
Human Peripheral Blood Stem Cells, Mesenchymal Stem Cells, Hematopoietic Stem Cells, Embryonic Stem Cells, Neural Stem Cells

1. Introduction
Stem cells are uncommitted cells with the ability of self-renewing, differentiating and regenerating or with the ability to reconstruct any tissue within in vivo environment [1] . Stem cells have been extensively studied during the last four or five decades due to their potential in the development of innovative therapeutic strategies [1] . Overall, stem cells can be divided into two groups, embryonic and adult stem cells [1] . Pluripotent embryonic stem cells (ESCs) originate from the inner cell mass of the blastocyst stage during embryonic development. They are very multipurpose type cells and can differentiate into all cell types of the body [1] . Adult stem cells are less versatile and could be categorized as multipotent stem cells that include two major groups―mesen- chymal stem cells (MSCs) and haematopoietic stem cells (HSCs) [1] -[6] . One of the major obstacles for the therapeutic use of ESCs is related to ethical regulation, as well as immunological incompatibilities that may rise up to uncontrolled development of malignancies or teratomas [1] . Conversely, the use of MSCs is free of such ethical concerns and requires less stringent criteria for transplantation [1] . MSCs are stem cells of stromal origin characterized by their multipotency with the ability to self-renew [2] [3] and differentiate to diverse cell phenotypes such as osteoblasts, chondrocytes, adipocytes, fibroblasts, tenocytes [4] [5] , hepatocytes, neural cells and/or epithelial cells [6] . Due to their crucial regenerative, reparative, angiogenic and immunomodulatory properties, MSCs have generated increased interest in a variety of biomedical disciplines and several areas of experimental and clinical applications [2] . Surely, marrow MSCs (BM MSCs) have been the most widely studied; however MSCs can be also isolated, with similar but not identical features, from different tissues including umbilical blood cord (UCB MSCs), placenta, adipose tissue (AT MSCs), trabecular bone, dental pulp and peripheral blood (PB MSCs) [7] -[12] . However, the limitation in the number of totally characterized autologous MSCs available may represent a major impediment to their use for adult stem cell therapy [13] . Furthermore, the in vivo use requires an extensive manipulation in vitro that eventually may compromise the quality, effectiveness and safety of the cells [13] . Multipotent HSCs are by definition the core of the entire repertoire of the haematopoietic lineages in vivo, maintaining the production throughout life [1] . HSCs are being used for the restoration of lympho-haematopoietic function after myeloablative, near myeloablative or non-myeloablative treatment [1] . The best-known location for HSCs is bone marrow, and bone marrow transplantation has become synonymous with hematopoietic cell transplantation [14] . Yet, bone marrow itself is increasingly infrequently used as a source due to an invasive harvesting procedure that requires general anesthesia. The transplantation also requires a strict HLA cross-matching compatibility between donor and recipient [14] . Therefore, there are definite advantages to using PBSCs as no ethic obligations are needed; PBSCs are readily available and are less exposed to immunologic challenges, and can be harvested with very low risk to the donor [13] . In addition, there is no need for a prolonged culture in vitro and they are able to differentiate to diverse cell phenotypes as their counterpart from other sources [13] .
The main aim of this study was to determine the presence of different sub-groups of pluripotent and multipotent stem cells present within human PB pool which, in the near future, may represent a breakthrough in the field of regenerative medicine and in the treatment of degenerative diseases. Reverse transcription polymerase chain reaction (RT PCR) was used to identify the expression of multipotent markers (Oct4, Sox2, OCN, Nestin, Nanog and DMP). Flowcytometry analysis confirmed that both adherent and non-adherent mononucleated cells were positive for a panel of mutipotency and pluripotency markers such as CD44, CD73, CD90, CD133, CD34, CD45, CD14, Nestin, SSEA3 and Tra1. Also quantification was the presence of 14 hormones in extracellular matrix components into the stem cell culture supernatants; additionally assessment was the spontaneous ability of hPBSCs to secrete high-level pro-inflammatory cytokines TNFα and IL-6 and low-level pro-inflammatory cytokines IFNy and IL-2. The isolated adherent and suspension mononucleated cells were able to maintain their stem cell properties during in-vitro culture by retaining their capacity to proliferate and differentiate when exposed to the appropriate culture medium.
2. Material and Methods
2.1. Isolation of Human Mononucleated Cells
Mononucleated cells were isolated from human peripheral blood samples obtained from healthy consent donors and following the guidelines of Health Department Research Ethic Committee of Ho Chi Minh city―Vietnam (Helsinki Declaration). Mononucleated cells were isolated by density gradient centrifugation using Ficoll-Pa- que™ PLUS (GE Healthcare, Uppsala, Sweden). The blood samples (35 ml each) were carefully layered 1:2 on Ficoll-Paque and centrifuged at 300 g for 20 min at 20˚C. The mononucleated cell layer, 4 × 107, at the plasma- Ficoll interface, were aspired and was washed three times with phosphate buffered saline and cultured in 25T flasks with free serum medium containing 2% (v/v) penicillin-streptomycin at 37˚C in a humidified atmosphere containing 5% CO2 for a period of 1 to 21 days, where it was analyzed the secretion capacity of these stem cells of different molecules as, cytokines, interleukines and hormones. After 6 days, non-adherent (suspension) mononucleated cells were transferred into new flasks. Both adherent and suspension mononucleated cells were maintained in the complete medium with twice weekly medium exchange for 14 - 21 days in in-vitro culture before further use. Suspension and adherent mononucleated cells were cultured in free serum medium (FSM- Life, Technology-CTSTM-StemProR, Canada). For both suspension and adherent mononucleated cells, the trypan blue exclusion assay was used to observe the proliferation of the cells.
2.2. Reverse Transcription Polymerase Chain Reaction Amplification
RT-PCR analysis was performed to evaluate the presence of a set of genes, Nanog, Oct4, Sox2 Nestin, Osteocalcin (OCN) and DMP responsible of stem cell multi-potency. Briefly, the total RNA of the adherent and suspension mono-nucleated cells that had been cultured in free serum medium was extracted using TRIZOL-LS (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The cDNA synthesis of total extracted RNA was carried out by using the iScript cDNA synthesis kit provided by Biorad (Biorad, Hercules, CA, USA). The cDNA synthesis was done by adding 15 µl of the extracted RNA into a thin wall tube containing 4 µl of the 5× iScript reaction mix and 1 µl of the iScript reverse transcriptase enzyme; after that, the cDNA synthesis was run in 5 min at 25˚C for annealing of the random hexamer and oligo (dT) to the RNA template, 30 min at 42˚C for the cDNA synthesis, and 5 min at 85˚C for inactivating the reverse transcriptase enzyme. After the cDNA synthesis, the multiplex PCR using specific primers (sequences listed in Table 1) were used to amplify and identify the cDNA that were reverse trancripted from the mRNA originated from Oct4, Sox2, OCN, Nestin, Nanog and DMP gene. The multiplex PCR were prepared from the multiplex PCR master mix (Qiagen, Hilden, Germany) added with the specific primers at the concentration of 0.5 µM for each. Four 4 multiplex PCR were prepared to detect OCT4 and GAPDH (plex 1); GAPDH, OCN and Nestin (plex 2); GAPDH, SOC2 and DMP (plex 3); GAPDH and NANOG (plex 4). The GAPDH were used in all 4 multiplex PCR as the internal control for the whole processing of the RT-PCR. The reaction volume of the multiplex PCR is 20 µl after adding 5 µl of cDNA, and the PCR was run in the MycyclerTM thermal cycler of Biorad with the thermo-cycling program of 95˚C for 15 min to activate the hot-start taq polymerase; follow with 40 cycles of 94˚C for 30 sec, 57˚C for 1

Table 1. List of the primers and sequences used for the RT-PCR to identify the expression of pluripotency markers (Oct4, Sox2, OCN, Nestin, Nanog and DMP).
min and 72˚C for 1 min; and terminate with 72˚C for 5 min. The PCR products were analyzed by agarose gel electrophoresis using GelRed (Biotinum, Hyward, CA, USA) as the fluorescent staining dye. In order to be sure that the PCR products are amplified from the mRNA of the target genes, the specific bands from the electrophoresis gel were cut and eluted, then carried-out the direct sequencing for the PCR product >250 bps (Nestin and Nanog) and sequencing after cloning to pGEM®-T Easy for the PCR products <250 bps (OTC4, SOC2, OCN and DMP).
2.3. Immunophenotyping by Flowcytometry Analysis
Immunephenotyping by flowcytometry analysis was performed to evaluate the presence of a set of markers CD44, CD73, CD90, CD133, CD 34, CD45, CD14, Nestin, SSEA3 and Tra1. Cell samples were washed twice in Dulbecco’s PBS containing 1% BSA (Sigma-Aldrich). Cells were stained for 30 minutes at 4˚C with anti- CD14-fluorescein isothiocyanate, anti-CD34-fluorescein isothiocyanate, anti-CD133-fluorescein isothiocyanate, anti-CD44-phycoerythrin, anti-CD45-fluorescein isothiocyanate, anti-CD90-phycoerythrin, anti-CD105-fluo- rescein isothiocyanate mAb, anti-nestin, anti-Tra1, anti-SSEA3 (BD Biosciences, Franklin Lakes, NJ, USA). Stained cells were analyzed by a FACSCalibur flow cytometer (BD Biosciences). Isotype controls were used for all analyses.
2.4. Immune Fluorescence Analysis SSEA3 and CD44 (Red Stain)
Immune fluorescence analysis was performed to evaluate the presence of SSEA3 and CD44 markers. Coat coverslips were prepared with polyethylineimine or poly-L-lysine for 1 hr at room temperature and rinsed with sterile H2O (3 × 5 minutes each) and sterilized them under UV light for at least 4 hrs. Cells were positioned on glass coverslips and rinsed in phosphate buffered saline (PBS). Samples were fixed either in 3% - 4% paraformaldehyde in PBS pH 7.4 for 15 min at room temperature and washed twice with ice cold PBS. Samples were then incubated for 10 minutes in PBS containing 0.25% Triton X-100 (or 100 μM digitonin or 0.5% saponin) and subsequently washed in PBS 3 × 5 minutes. The sample was incubated with 1% BSA in PBST for 30 min to block unspecific binding of the antibodies, cells were then incubated with antibody for 1 hour at room temperature. Cells were then decanted and washed with PBS 3 × 5 minutes. Successively, cells were incubated with the secondary antibody in 1% BSA for 1 hr at room temperature in the dark; the secondary antibody solution was decanted and washed with PBS 3 × 5 minutes in the dark. In the following step, cells were incubated with 0.1 - 1 μg/ml Hoechst or DAPI (DNA stain) for 1 min and rinsed with PBS. In mount procedure all coverslip were sealed with nail polish to prevent drying and movement under microscope and were stored in the dark at −20˚C or +4˚C.
2.5. Cytokine Quantification
We quantified cell supernatant concentration of 4 different extracellular matrix-related proteins: pro-inflamma- tory cytokines and interleukins TNFα, IFNy, IL-2 and IL-6. Commercial Luminex kits were used: Human Cytokine 30-plex Assay (Invitrogen, Carlsbad, California, USA), Fluorokine MAP TGF-β Multiplex Kit (R&D, Minneapolis, Minnesota, USA). Procedures were performed according to the manufacturers’ instructions.
2.6. Hormone Quantitative Analysis
We quantified concentration of a panel of 14 different extracellular matrix-related hormones: Commercial Elecsys reagent kits were used (Roche Diagnostics―USA) and run by Cobas 6000 (Roche Diagnostics―USA). 10 - 40 µl samples were used in the presence of a biotinylated monoclonal specific antibody. The reaction mixture was aspirated into the measuring cell where the microparticles are magnetically captured onto the surface of the electrode. Unbound substances were then removed with ProCell/M (Roche Diagnostic―USA). Application of a voltage to the electrode then induces chemiluminescent emission, which is measured by a photomultiplier. Results were determined via a calibration curve, which is the instrument specifically generated by 2-point calibration and a master curve provided via the reagent barcode.
2.7. Cytochemical Staining of Osteoblasts Derived from hPBSCs
Induction of osteoblastic differentiation was performed by using a “ready to go” osteogenic medium containing basal medium, osteogenic stimulatory supplement, dexamethasone, ascorbic acid, beta glycerol phosphate (Stem Cells―Canada). Once the adherent cells reached a confluence of 90% a medium with supernatants was removed and new osteogenic medium was added. Meanwhile supernatants were isolated by centrifugation (3000 rpm) and immerged in a new flask with osteogenic medium. Mineral matrix deposits and bone nodules of osteoblasts, from hPBSCS adherent and supernatant cells were evaluated by staining cell cultures by alizarin red (AR) and alkaline phosphatase (AP).
2.7.1. Alizarin Red Stain Procedure
The presence of calcium deposits were detected by washing cells with cold PBS and fixing them in 10% NFB-neutral formalin buffer solution for 30 minutes in chemical hood. Cells were rinsed 3 times with distilled water and immerged in 2% (w/v) solution of alizarin red for 30 seconds to 5 minutes. Cells were rinsed again with distilled water for 2 - 3 times and checked under inverse microscope and photographed.
2.7.2. Alkaline Phosphatase Stain Procedure
The presence of alkaline phosphates was detected by washing cells with cold PBS and fixing them in 10% NFB-neutral formalin buffer solution for 30 minutes in chemical hood and stained with solution naphtol As-MX-PO4 (Sigma) and Fast red violet LB salt (Sigma) for 45 minutes in dark at room temperature. Cells were rinsed three times with distilled water and checked by inverse microscope and photographed.
2.8. Cell Count by Hemocytometer
The formula is c = n/v where c is cell concentration (cells/ml). It has been used Neubauer slides, the depth of chamber is 0.1 mm and it has been used the central chamber (1 mm2), and v is 1 × 10−4, therefore the final formula is c = n × 10−4.
2.9. Statistical Analysis
Data were statistically analyzed using paired t-tests. Effects were considered statistically significant at p < 0.05.
3. Results
3.1. Isolation of Mononucleated Cells
Human periheral blood mononucleated cells consisted of miscellaneous undifferentiated sub-sets of stem cells composed of MSCs, HSCs, NSCs ESCs and their progenitors, along with leukocytes, dendric cells and macrophages. Mononucleated cells were isolated from the buffy coat of peripheral blood using a density gradient centrifugation on Ficoll-Paque where differences in density separated mononucleated cells from other blood cells. In this study, we further separated the mononucleated cells according to their physical characteristics. Non-ad- herent (suspension) and adherent cells were cultured separately after 6 days of isolation. Both adherent and suspension mononucleated cells were maintained in the serum free medium (SFM) for 14 - 18 days of culture for the purpose of separating adherent and suspension cells and also to deplete most of the unwanted cells that have a short lifespan. Suspension cells without staining appear morphologically rounded; MSCs are cells that adhere to the tissue culture plastic, tended to appear around day 4 - 6 as well and displayed a fibroblastic-like appearance (Figure 1(b) & Figure 1(c)); NSCs adherent cells appeared by the end of first week, displaying different morphologies typical of variegate neural cell family such as neuron precursor cells which generate neurons but not glia, early elongated attached neuron progenitor, oligodendrocyte precursors and branched fibrous astrocytes (Figures 2(a)-(d)); later on at day 4 - 6 floating embryonic like cells were present with a typical grape shape like which eventually part of them adhered and amalgamated into a unique mass with rough extra-cellular surface (Figures 3(a)-(c)); the morphology of the earlier progenitor HSCs included a large single nucleus that occupied much of the cytoplasmic space, resembling that of lymphocytes (Figure 4(a) & Figure 4(b)).
3.2. Characterization of Pluripotency and Multipotency of Human Peripheral Blood Stem Cells
Morphology alone is not sufficient to prove that HSCs, MSCs, ESCs and NSCs are present in hPB mononucleated


 (a) (b) (c)
(a) (b) (c)
Figure 1. Inverted phase-contrast micrographs showing the morphologies of primary and second generation peripheral blood-derived like mesenchymal stem cells cultured for 5 days (a) and 7 days (b) and 10 days (c) (original magnification ×100- 200-400).



 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 2. Inverted phase-contrast micrographs showing the morphologies of primary and second generation peripheral blood-derived like neuronal progenitor stem cells cultured for 15 days (a)-(d) (original magnification ×200).


 (a) (b) (c)
(a) (b) (c)
Figure 3. Inverted phase-contrast micrographs showing the morphologies of primary and second generation peripheral blood-derived like embryonic stem cells cultured for 5 to 10 days (a)-(c) (original magnification ×200-400).
cells. In the PB-SSCs cultures, approximately 0.5 - 1 million surface adherent cells were obtainable from each sample flask obtained from 35 ml fresh blood, after 2 weeks from cell seeding (passage 0). Though there still is a controversy among scientist about the specificity of markers and cell phenotype, the existence of these sub-sets of stem cells with multipotent-pluripotent phenotype was further characterized by RT-PCR, flowcytometry analysis and fluorescence analysis. An empirical but not conclusive division was therefore made, flowcytometry results showed adherent mononucleated cells CD14, CD34 and CD45 for human hematopoietic lineage; CD44, CD73, CD90 and CD105 for human mesenchymal lineage; CD133 and Nestin for human neural lineage [15] ; SSEA3 and Tra1 for human embryonic lineage (Graph 1). Growth curve of primary cultured human peripheral- blood derived stem cells, both adherent and suspended cells is shown, in the graphic cell number showed a gradual

 (a) (b)
(a) (b)
Figure 4. Inverted phase-contrast micrographs showing the morphologies of primary and second generation peripheral blood-derived like hematopoietic stem cells cultured for 7 - 10 days (a) (b) (original magnification ×400).
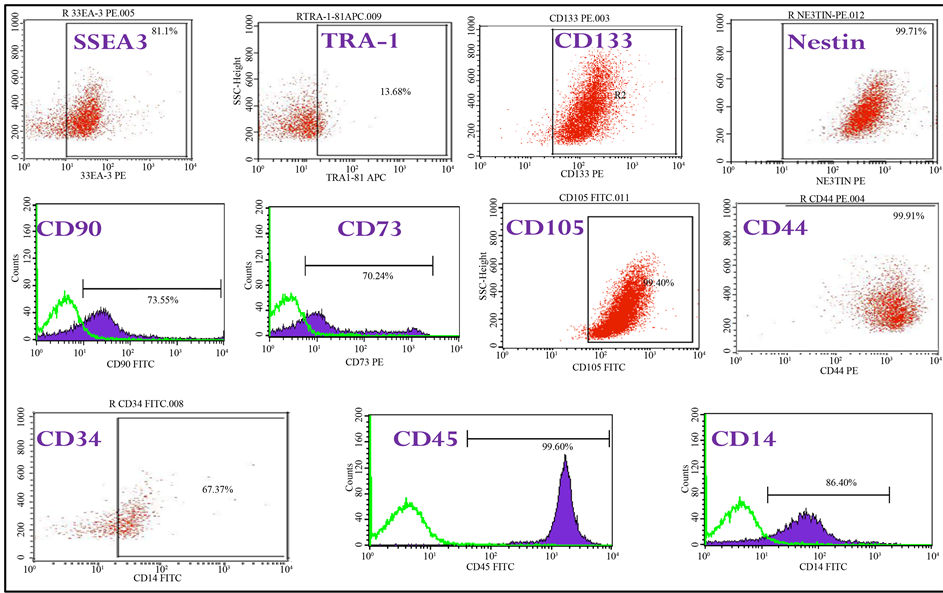
Graph 1. Immunophenotype of hPBSCs isolated by fibrin microbeads (FMB). FACS analysis of the immunophenotype profile have been carried out for hematopoietic marker CD45, CD34 and CD14, for mesenchymal antigens CD90, CD73, CD44 and CD105, for neural stem cell antigen CD133 and Nestin, for embryonic stem cell antigens SSEA3 and TRA1 of the initial peripheral blood progenitor cell sample before isolation. Cells isolated by FMB technique were harvested 18 - 19 days following harvesting and cultured for further passages. Shaded histograms represent the fluorescenc from control cells stained with only second antibody; non-filled histograms represent positive staining of cells with the indicated antibody.
inflection during the first days of culture and raised soon after (Graph 2). Figure 5 indicates the cell membrane localisation of anti-SSEA3 protein in hPBSCs expressing the pluripotent marker SSEA-3 (red fluorescence); and this figure also demonstrates the cell membrane localisation of CD44 by staining with an anti-CD44 monoclonal antibody and Rhodamine conjugate (red fluorescence) in hPBSCs. As shown in Figure 6, the amplification of the RT-PCR products of GAPDH and a panel of transcription factors that regulate self-renewal and pluripotency in hESCs, Oct4, Sox2, OCN, Nestin, Nanog and DMP [16] , and the results of sequencing of the amplified
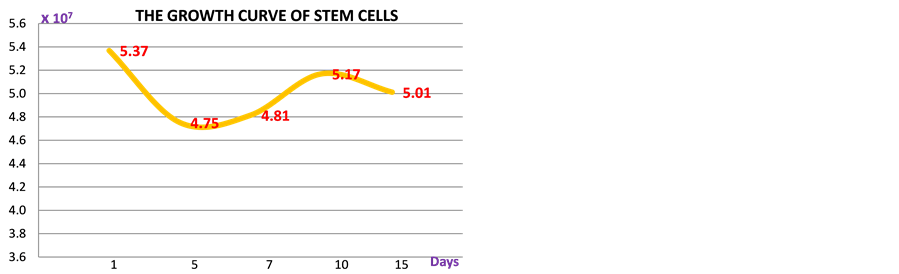
Graph 2. Growth curve of primary cultured human peripheral- blood derived stem cells, both adherent and suspended cells. The cell number showed a gradual inflection during the first days of culture and raised soon after. Though the growth rate seemed to be steady stem cells showed to maintain their differentiation and proliferation capacity until the third generation.

 (a) (b)
(a) (b)
Figure 5. (a) Cell membrane localisation of anti-SSEA3 protein in hPBSCs expressing the pluripotent marker SSEA-3 (red fluorescence); (b) Cell membrane localisation of CD44 was stained with an anti-CD44 monoclonal antibody and rhodamine conjugate (red fluorescence) in hPBSCs.
 Plex1 Plex2 Plex3 Plex4
Plex1 Plex2 Plex3 Plex4
Figure 6. Reverse transcription polymerase chain reactions (RT- PCRs) were performed on both adherent and suspended mononucleated cells. GAPDH and Sox2, Oct4, Nanog, Nestin, Dmp and osteocalcin (Ocn) were expressed in both types of mononucleated cells. A 1000-bp DNA size marker was used to identify the approximate size of the RT-PCR products and GAPDH was used as internal control. Plex1 indicates the expression of GAPDH 94 bps, OCT4 144 bps; Plex2 indicates the expression of GAPDH 94 bps, OCN 150 bps, Nestin 496 bps; Plex3 indicates the expression of GAPDH 94 bps, Sox2 151 bps, DMP 200 bps; and Plex4 indicates the expression of GAPDH 94 bps, Nanog 346 bps.
products shown the Table 2 did confirm all come from the mRNA of the target genes.
3.3. Induction to Osteoblast Lineage
Morphology changes were detected after 7 days in osteogenic differentiation medium. Viability cells decreased, and the cells morphologically from their original fibroblast-like shape changed to a polygonal appearance. At second week into the experiment, 4 changes of osteogenic differentiation medium was performed, the cell shape became flat and started to create compacted clusters and were observed the presence of typical mineralized nodules. 21 days into the experiment, the number of mineralized nodules increased, the cells developed into typical cubic shape, at this stage the density of mineralized nodules was relatively stable, suggesting they had reached a rather stable differentiated condition. The mineralized nodules were stained positively dark red color by alizarin red and violet by alkaline phopshatase (Figure 7), confirming that the human PBSCs had eventually differentiated toward osteoblast phenotype.
4. Discussion
Proliferation of Mononucleated Cells
Stem cells possess two properties: the ability to self-renew or proliferate and the ability to differentiate into a vast repertoire of specialized cells; therefore the use of cell therapy for the repair of damaged tissues as bones, cartilage or skin with or without the support of scaffolds is well established, demonstrating good short to medium
Table 2. Complete sequencing from PCR of OCT4, OCN, Nestin, Sox2, DMP and Nanog.

 (a) (b)
(a) (b)
Figure 7. Osteoblast like cells from hPBSCs, adherent cells at 21 days were directly induced by pouring osteogenic medium (Gibco USA) to osteoblast cell line, cells stained positive for both Alizarin red (a) and Alkaline phosphatase (b) ×200.
term outcomes either in vitro of in vivo [17] -[21] . However, it is clear the presence of inherent limitations associated with current procedures include the low cell yield from the tissues obtained, the invasiveness of the first stage of the procedure required to obtain the needed amount of tissue and, the site morbidity associated with the procedure [13] [17] . Thus, understanding the human SCs in vivo dynamic micro-environment, certainly will help to develop a better and safer procedure in clinical application [17] [22] [23] . Numerous studies have emphasized the importance of the niche as places where constant networks take place between stem cells and neighboring cells, secreted factors, inflammation and scarring, extracellular matrix, physical parameters such as shear stress and tissue stiffness and environmental signals such as hypoxia [24] . In many adult tissues, the stem cell niche contains a variety of cell types, each with a distinct function [24] . This is evident in the hematopoietic microenvironment localized in the marrow space, in peripheral blood or in adult bone and skin and include a range of different cell types such as progenitor stem cells, embryonic like cells, vascular and neural cells, megakaryocytes, macrophages and immune cells―each have important roles and can be considered to define distinct SC niches [17] [19] [24] [25] . Communication between stem cells and niche cells takes place through physical interactions, or indirectly through secreted factors as cytokines, interleukins or growth factors that mediate communication between distant cells [25] . For instance, mobilization of SCs from their niche either from bone marrow or peripheral blood is performed by using cytokines such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF) or interleukin growth factor 1 (IGF1) to provide treatment in hematological malignancy, bone marrow failure and rare genetic disorders [25] [26] . Therefore, the key trait of tissue regeneration can only be clarified by unveiling this complex coordination among all this components and their intimate capacity of communicate each other in vivo [25] .
Identification procedure for SCs is still an open debate, morphological analysis are not sufficient as some stem cells such as HSCs might be morphologically similar to white blood cells and may carry same molecular patterns [27] . Therefore, cell surface markers and cluster of differentiation antigens (CD) became a standard tool to characterize the SC subpopulations [27] . It is also widely confirmed that cell surface markers on SCs in adult bone marrow kinetically modulate in relation to the cell-cycle position and the differentiating behavior [27] . Hence, in this study, the isolated stem cells HSCs, ESCs (suspension) and MSCs, NSCs (adherent) as components were further characterized by RT-PCR, flowcytometry analysis and fluorescence analysis during a period of 1 to 21 days. Table 3 & Table 4 show that our cells secrete different quantity of molecules and hormones during a period of 21 days. It was established the expression of pluripotent markers such as Oct4, Sox2, OCN, Nestin, Nanog and DMP and together with these data, our results showed that the outgrowth of hPB SCs included type of cells present in the whole collection and the cell density for seeding probably influenced the clone formation; sparsely distributed cells were not observed after 7 days of culture. Nonetheless, we are aware of few existing issues related to the specificity of some markers, as several markers are associated with more than one stem cell lineage; for instance CD133 that can be also used as either hematopoietic stem cell or embryonic-neural stem cell precursor markers or, CD 90 that can be expressed by both embryonic and mesenchymal
Table 3. Human PBSCs at day 15 were tested for hormone secretion, cells were divided on two group from female and male donors. As the results clearly showed there is a very deep difference between the two groups especially in relation of androgen hormone secretion.
Table 4. Human PBSCs were tested at day 1-5-7-10-18-21 for cytokine and interleukines TNFα, INFγ, IL2 and IL6. Secretion steadily increased during the first 18 days to slow down by the 21st day. As the results clearly showed cells are capable to secrete high level of IL6 and TNFα and low level of IL2 and INFγ.
stem cells [28] -[32] . An additional challenge is that markers that identify SCs may also be used as predictor of tumor stem cells, generating an extra difficulty in distinguishing them from tumor SCs [33] . For this reason, we provided an empirical but not conclusive categorization, where CD14, CD34 and CD45 indicated human hematopoietic lineage; CD44, CD73, CD90 and CD105 indicated human mesenchymal lineage; TRA1 and SSEA3 indicated embryonic lineage and Nestin and CD133 neural stem cell lineage [16] . Of note, the outgrowth of different types of stem cells, MSCs, NSCs, HMSCs, ESCs and, their amount in vitro was variable, somehow in relation with the condition of the donor. In fact, we noticed that in vitro cultured cells from donors suffering of neural degenerative disease (multiple sclerosis-MS), cells spontaneously differentiated to higher quantity of neural progenitors cells compared to healthy donor cells (data not shown). We speculate that these results may indicate that both original molecular environment and the cell-cell interaction play a more important role than the paracrine effect in inhibiting or promoting the differentiation of cell-derived population. This mechanism would probably remain a homeostasis in peripheral blood that a little stem cell population circulates under normal condition, and only when monocytes are activated and consumed in the regeneration and repair stem cells are called in and start to proliferate and differentiate [22] [34] [35] . In this study, the proliferation of suspension and adherent mononucleated cells in an in-vitro culture was investigated at different days of culture, using the trypan blue exclusion assay respectively. These results show that suspension and adherent mononucleated cells were able to proliferate during in-vitro culture conditions. Compare with other type of stem cell sources, hPBSCs showed to have different morphology and possess some practical advantages. They exist in larger numbers in the human body than bone marrow stromal stem cells or adipose-derived stromal stem cells, and they can be obtained with relative ease procedure and without causing serious harm to the donor [36] . For instance, unlike hBM-MSCs, hPB-MSCs revealed to have a greater variety of adult cells, a higher number of rounded shaped cells than those in BM-MSCs and a reasonable number of pure stem cells. Eventually, one of the most interesting findings in this study is that hPB derived SCs are composed by four different types of stem cells diverse in morphology, phenotypes and gene expression. This is one of the fewest researches categorizing pluripotent and multipotent stem cells progenitors in human peripheral blood with hematopoietic, neural, mesenchymal and embryonic phenotype features, which extends the traditional understanding about the biology of stem cells system. As a recent result on hHSC merged into the field of immunology, with new findings emphasized the role played by immunologic signals (such as IFN-α, IFN-γ, or TNF-α) in the regulation of the HSC compartment [37] . In this article, we have unveiled a previously unsuspected ability of hPBSCs to release a specific variety of pro-inflammatory molecules such as IFN-γ, TNF-α, IL2 and IL6 over a period of 18 days and several types of hormones such as testosterone, progesterone, estrogen, DHEA, IGF1, TSH, T3-4, leptin, FSH, LH, ACTH and cortisol. Of note, proliferation of primary suspension and adherent mononucleated cells from day 1 and day 15 showed no significant increase in viable cell numbers in both cell populations. Nevertheless, isolated stem cells amongst adherent and suspension mononucleated cells were still able to maintain their stem cell properties during in-vitro culture by retaining their capacity to self-renew or proliferate and differentiate into specialized cells. Human PB derived SCs cultured in osteogenic medium differentiated into osteoblasts and produced mineralized extracellular matrix, showing that hPBSCs may have favorable properties for bone tissue engineering. Though, hPBSCs in vitro culture did not show to replicate as much as their counter part from BM, AT or UCB, therapies based on peripheral blood stem cell harvests are often life-saving [1] [17] yet is well established that, stem cells derived from bone marrow, embryonic and adipose tissues undergo spontaneous malicious alteration during long-term in vitro culture [36] [38] [39] . However, to avoid any malignant complication due to long term in vitro culturing we have planned to use PB primary stem cells that undergo only for a short period of culture, within 7 to 18 days. In addition, the use of hPBSCs does not present any ethical controversial as the embryo-derived mesenchymal stem cells may do [36] [38] [39] . Eventually, this study suggests that hPBSCs offer advantages in availability, biological activities, cell adhesion, immunogenecity, cytokine production, hormone secretion and osteoblastic differentiation. The combination of these favorable properties may make hPBSCs a promising candidate for tissue engineering and degenerative condition.
5. Conclusions
To conclude, blood reveals to be a critical site where different types of stem cell population with multipotent and pluripotent capabilities can be found, representing an extremely useful source for clinical applications. Although certain divergences within the peripheral blood SCs literature result in differing descriptions of their biological properties, these variances may be explicable by the fact that these distinct populations express some dissimilarity in function and number closely-related from patient to patient. In summary, blood SCs can self-renew to a certain extent and can differentiate. It is more interesting that, they can display a variety of significant cell functions including repair functions within damaged area and are able to support appropriate cell and tissue renewal to replace the injured tissues. It appears that these stem cells work together in a very interactive manner, almost supporting each other in any activity that they are involved with. Concomitantly, blood SCs in our study in vivo showed to have exerted an immune-modulatory function similar to MSCs and no modification or rejection of blood SC after allogeneic human transplantations has been observed to date to our patients (data not shown). Results indicate an enormous potential for the clinical use of these cells in the field of regenerative medicine.
Therefore, in contrast to the practical and ethical limitations of bone marrow, adipose tissue, umbilical cord and embryonic tissue, SC from peripheral blood can be obtained in larger amounts, and the required numbers of these stem cells can be transplanted for tissue replacement and for regenerative purposes.
Cite this paper
CiroGargiulo,Van HungPham,NguyenThuy Hai,Kieu C. D.Nguyen,Pham VanPhuc,KenjiAbe,VeronicaFlores,MelvinShiffman, (2015) Isolation and Characterization of Multipotent and Pluripotent Stem Cells from Human Peripheral Blood. Stem Cell Discovery,05,19-32. doi: 10.4236/scd.2015.53003
References
- 1. Ab Kadir, R., Hisham Zainal Ariffin, S., Megat Abdul Wahab, R. and Senafi, S. (2012) Molecular Characterisation of Human Peripheral Blood Stem Cells. South African Journal of Science, 108, 1-7.
http://dx.doi.org/10.4102/sajs.v108i5/6.939 - 2. Trivanovic, D., Kocic, J., Mojsilovic, S., Krstic, A., Ilic, V., Djordjevic, I.O., Santibanez, J.F., et al. (2013) Mesenchymal Stem Cells from Peripheral Blood and Umbilical Cord Wharton’s Jelly. Srpski arhiv za celokupno lekarstvo, 141, 178-186.
http://dx.doi.org/10.2298/SARH1304178T - 3. Bianco, P., Riminucci, M., Gronthos, S. and Robey, P.G. (2001) Bone Marrow Stromalcells: Nature, Biology, and Potential Applications. Stem Cells, 19, 180-192.
http://dx.doi.org/10.1634/stemcells.19-3-180 - 4. Caplan, A.I. (2007) Adult Mesenchymal Stem Cells for Tissue Engineering versus Regenerative Medicine. Journal of Cellular Physiology, 213, 341-347.
http://dx.doi.org/10.1002/jcp.21200 - 5. Chamberlain, G., Fox, J., Ashton, B. and Middleton, J. (2007) Mesenchymal Stem Cells: Their Phenotype, Differentiation Capacity, Immunological Features, and Potential for Homing. Stem Cells, 25, 2739-2749.
http://dx.doi.org/10.1634/stemcells.2007-0197 - 6. Gimble, J.M., Guilak, F., Nuttall, M.E., Sathishkumar, S., Vidal, M. and Bunnell, B.A. (2008) In Vitro Differentiation Potential of Mesenchymal Stem Cells. Transfusion Medicine and Hemotherapy, 35, 228-238.
http://dx.doi.org/10.1159/000124281 - 7. Wagner, W., Wein, F., Seckinger, A., Frankhauser, M., Wirkner, U., Krause, U., et al. (2005) Comparative Characteristics of Mesenchymal Stem Cells from Human Bone Marrow, Adipose Tissue, and Umbilical Cord Blood. Experimental Hematology, 33, 1402-1416.
http://dx.doi.org/10.1016/j.exphem.2005.07.003 - 8. Kassis, I., Zangi, L., Rivkin, R., Levdansky, L., Samuel, S., Marx, G., et al. (2006) Isolation of Mesenchymal Stem Cells from G-CSF-Mobilized Human Peripheral Blood Using Fibrin Microbeads. Bone Marrow Transplant, 37, 967-976.
http://dx.doi.org/10.1038/sj.bmt.1705358 - 9. Sakaguchi, Y., Sekiya, I., Yaqishita, K. and Muneta, T. (2005) Comparison of human Stem Cells Derived from Various Mesenchymal Tissues. Arthritis & Rheumatism, 52, 2521-2529.
http://dx.doi.org/10.1002/art.21212 - 10. Gronthos, S., Mankani, M., Brahim, J., Robey, P.G. and Shi, S. (2000) Postnatal Human Dental Pulp Stem Cells (DPSCs) in Vitro and in Vivo. Proceedings of the National Academy of Sciences of the United States of America, 97, 25-30.
http://dx.doi.org/10.1073/pnas.240309797 - 11. Wang, H.S., Hung, S.C., Peng, S.T., Huang, C.C., Wei, H.M., Guo, Y.J., et al. (2004) Mesenchymal Stem Cells in the Wharton’s Jelly Umbilical Cord. Stem Cells, 22, 1330-1337.
http://dx.doi.org/10.1634/stemcells.2004-0013 - 12. Locke, M., Feisst, V. and Dunbar, P.R. (2011) Concise Review. Human Adipose Derived Stem Cells: Separating Promise from Clinical Need. Stem Cells, 29, 404-411.
http://dx.doi.org/10.1002/stem.593 - 13. Patel, A.N. and Genovese, J. (2011) Potential Clinical Applications of Adult Human Mesenchymal Stem Cell (Prochymal®) Therapy. Stem Cells and Cloning: Advances and Applications, 4, 61-72.
- 14. Domen, J., Wagers, A. and Weissman, I.L. (2006) Regenerative Medicine Bone Marrow (Hematopoietic) Stem Cells. National Institute of Health, 2, 14-28.
- 15. Sun, Y., Kong, W., Falk, A., Hu, J., Zhou, L., Pollard, S. et al. (2009) CD133 (Prominin) Negative Human Neural Stem Cells Are Clonogenic and Tripotent. PLoS ONE, 4, e5498.
http://dx.doi.org/10.1371/journal.pone.0005498 - 16. Callas, A., Pook, M., Treika, A. and Maimets, T. (2014) SOX2 Is Regulated Differently from NANOG and OCT4 in Human Embryonic Stem Cells during Early Differentiation Initiated with Sodium Butyrate. Stem Cells International, 2014, Article ID: 298163.
http://dx.doi.org/10.1155/2014/298163 - 17. Chong, P.P., Selvaratnam, L., Abbas, A.A. and Kamarul, T. (2011) Human Peripheral Blood Derived Mesenchymal Stem Cellsdemonstrate Similar Characteristics and Chondrogenic Differentiation Potential to Bone Marrow Derived Mesenchymal Stem Cells. Journal of Orthopedic Research, 30, 634-642.
- 18. Aubin, J.E. and Heersche, J.N. (2000) Osteoprogenitor Cell Differentiation to Mature Bone Forming Osteoblasts. Drug Development Research, 49, 206-215.
http://dx.doi.org/10.1002/(SICI)1098-2299(200003)49:3<206::AID-DDR11>3.0.CO;2-G - 19. Undale, A.H., Westerndorf, J.J., Yaszemski, M.J. and Khosla, S. (2009) Mesenchymal Stem Cells for Bone Repair and Metabolic Bone Disease. Mayo Clinic Proceedings, 84, 893-902.
http://dx.doi.org/10.4065/84.10.893 - 20. Aubin, J.E., Malaval, F. and Gupta, A.K. (1995) Osteoblasts and Chondroblasts Differentiation. Bone, 17, 77-83.
http://dx.doi.org/10.1016/8756-3282(95)00183-E - 21. Homicz, M.R., Schumacher, B.L., Sah, R.L. and Watson, D. (2012) Effects of Serial Expansion of Septal Chondrocytes on Tissue-Engineered Neocartilage Composition. Otolaryngology Head and Neck Surgery, 127, 398-408.
http://dx.doi.org/10.1067/mhn.2002.129730 - 22. Gang, H., Peng, L., Jie, F. and Yan, J. (2011) A Novel Population of Mesenchymal Progenitors with Hematopoietic Potential Originated from CD14‾ Peripheral Blood Mononuclear Cells. International Journal of Medical Sciences, 8, 16-29.
- 23. Grage-Griebenow, E., Flad, H.D. and Ernst, M. (2001) Heterogeneity of Human Peripheral Blood Monocyte Subsets. Journal of Leukocyte Biology, 69, 11-20.
- 24. Lane, L.W., Williams, D.A. and Watt, F.M. (2014) Mod-ulating the Stem Cell Niche for Tissue Regeneration. Nature Biotechnology, 32, 795-803.
http://dx.doi.org/10.1038/nbt.2978 - 25. Havens, A.M., Shiozawa, Y., Jung, Y., Sun, H., Wang, J., McGee, S., et al. (2012) Human Very Small Embryonic-Like Cellsgenerate Skeletal Structures, in Vivo. Stem Cells and Development, 1, 1-9.
- 26. Haider, H.K., Jiang, S., Idris, N.M. and Ashraf, M. (2008) IGF-1-Overexpressing Mesenchymal Stem Cells Accelerate Bone Marrow Stem Cell Mobilization via Paracrine Activation of SDF-1α/CXCR4 Signaling to Promote Myocardial Repair. Circulation Research, 103, 1300-1308.
http://dx.doi.org/10.1161/CIRCRESAHA.108.186742 - 27. Senthil, P. (2013) Adult Stem Cell and Embryonic Stem Cell Markers. Mater Methods, 3, 200.
- 28. Zhao, W., Ji, X., Zhang, F., Li, L. and Ma, L. (2012) Embryonic Stem Cell Markers. Molecules, 17, 6196-6236.
http://dx.doi.org/10.3390/molecules17066196 - 29. Draper, J.S., Pigott, C., Thomson, J.A. and Andrews, P.W. (2002) Surface Antigens of Human Embryonic Stem Cells: Changes upon Differentiation in Culture. Journal of Anatomy, 200, 249-258.
http://dx.doi.org/10.1046/j.1469-7580.2002.00030.x - 30. Xu, C.H., Inokuma, M.S., Denham, J., Golds, K., Kundu, P., Gold, J.D., et al. (2001) Feeder-Free Growth of Undifferentiated Human Embryonic Stem Cells. Natural Biotechnology, 19, 971-974.
http://dx.doi.org/10.1038/nbt1001-971 - 31. Skottman, H., Mikkola, M., Lundin, K., Olsson, C., Strömberg, A.M., Tuuri, T., et al. (2005) Gene Expression Signatures of Seven Individual Human Embryonic Stem Cell Lines. Stem Cells, 23, 1343-1356.
http://dx.doi.org/10.1634/stemcells.2004-0341 - 32. Mingyu, Z., Tao, S., Liang, Y., Chen, R., Wu, L., Yang, Z., et al. (2008) Nestin and CD133; Valuable Stem Cell-Specific Markers for Determining Clinical Outcome of Glioma Patients. Journal of Experimental & Clinical Cancer Research, 27, 85-92.
http://dx.doi.org/10.1186/1756-9966-27-85 - 33. Knoepfler, P. (2009) Deconstructing Stem Cell Tumorigenicity: A Roadmap to Safe Regenerative Medicine. Stem Cells, 27, 1050-1056.
http://dx.doi.org/10.1002/stem.37 - 34. Schurch, C.M., Riether, C. and Ochsenbein, A.F. (2014) Cytotoxic CD8+ T Cells Stimulate Hematopoietic Progenitors by Promoting Cytokine Release from Bone Marrow Mesenchymal Stromal Cells. Cell Press, 14, 460-472.
http://dx.doi.org/10.1016/j.stem.2014.01.002 - 35. Weiner, R.S. and Kincade, P.W. (2014) 9-1-1: HSCs Respond to Emergency Calls. Cell Press, 14, 415-416.
- 36. Zhong, Z.N., Zhu, S.F., Yuan, A.D., Lu, G.H., He, Z.Y., Fa, Z.Q., et al. (2012) Potential of Placenta-Derived Mesenchymal Stem Cells as Seed Cells for Bone Tissue Engineering: Preliminary Study of Osteoblastic Differentiation and Immunogenicity. Orthopedics, 35, 779-788.
http://dx.doi.org/10.3928/01477447-20120822-07 - 37. Rossi, L., Salvestrini, V., Ferrari, D., Virgilio, F.D., Lemoli, R.M., et al. (2012) The Sixth Sense: Hematopoietic Stem Cells Detect Danger through Purinergic Signaling. Blood, 120, 2365-2375.
http://dx.doi.org/10.1182/blood-2012-04-422378 - 38. Rosland, G.V., Svendsen, A., Torsvik, A., Sobala, E., McCormack, E., Immervoll, H., et al. (2009) Long-Term Cultures of Bone Marrow-Derived Human Mesenchymal Stem Cells Frequently Undergo Spontaneous Malignant Transformation. Cancer Research, 69, 5331-5339.
http://dx.doi.org/10.1158/0008-5472.CAN-08-4630 - 39. Izadpanah, R., Kaushal, D., Kriedt, C., Tsien, F., Patel, B., Dufour, J., et al. (2008) Long-Term in Vitro Expansion Alters the Biology of Adult Mesenchymal Stem Cells. Cancer Research, 68, 4229-4238.
http://dx.doi.org/10.1158/0008-5472.CAN-07-5272
NOTES

*Corresponding author.